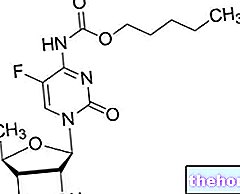
Aktivne sestavine: kapecitabin
Xeloda 150 mg filmsko obložene tablete
Paketni vložki Xeloda so na voljo za velikosti pakiranj:- Xeloda 150 mg filmsko obložene tablete
- Xeloda 500 mg filmsko obložene tablete
Zakaj se zdravilo Xeloda uporablja? Za kaj je to?
Zdravilo Xeloda spada v skupino zdravil, imenovanih „citostatična zdravila“, ki ustavijo rast rakavih celic. Zdravilo Xeloda vsebuje 150 mg kapecitabina, ki sam po sebi ni citostatično zdravilo. Šele ko ga telo absorbira, se spremeni v aktivno zdravilo proti raku (v večji meri v tumorskih tkivih kot v normalnih tkivih).
Zdravniki predpisujejo zdravilo Xeloda za zdravljenje raka debelega črevesa, danke, želodca ali dojk. Zdravilo Xeloda je predpisano tudi za preprečevanje pojava novega raka debelega črevesa po popolni kirurški odstranitvi tumorja.
Zdravilo Xeloda se lahko uporablja samostojno ali v kombinaciji z drugimi zdravili.
Kontraindikacije Kadar zdravila Xeloda ne smete uporabljati
Ne jemljite zdravila Xeloda:
- če ste alergični na kapecitabin ali katero koli sestavino tega zdravila (navedeno v poglavju 6). Zdravniku morate povedati, če veste, da ste alergični ali na to zdravilo pretirano reagirate,
- če ste imeli predhodno resno reakcijo na zdravljenje s fluoropirimidinom (skupina zdravil proti raku, kot je fluorouracil),
- če ste noseči ali dojite,
- če imate v krvi prenizko raven belih krvnih celic in trombocitov (levkopenija, nevtropenija ali trombocitopenija),
- če imate hude težave z jetri ali ledvicami,
- če imate znano pomanjkanje encima dihidropirimidin dehidrogenaze (DPD), ki sodeluje pri presnovi uracila in timina ali
- če se trenutno zdravite ali ste bili v zadnjih 4 tednih zdravljeni z brivudinom, sorivudinom ali snovmi podobnih razredov kot del terapije za herpes zoster (norice ali ogenj sv. Antona).
Previdnostni ukrepi pri uporabi Kaj morate vedeti, preden boste vzeli zdravilo Xeloda
Preden vzamete zdravilo Xeloda, se posvetujte s svojim zdravnikom ali farmacevtom:
- če imate bolezen ledvic ali jeter,
- če ste imeli ali imate težave s srcem (na primer nepravilen srčni utrip ali bolečine, ki sevajo od prsnega koša do čeljusti in obratno zaradi fizičnega napora in zaradi težav s pretokom krvi v srce),
- če imate možgansko bolezen (na primer tumor, ki se je razširil v možgane) ali okvaro živcev (nevropatija),
- če imate neravnovesje v ravni kalcija (zaznano pri preiskavah krvi),
- če imate sladkorno bolezen,
- če zaradi hude slabosti in bruhanja ne morete hraniti hrane ali vode v telesu,
- če imate drisko,
- če ste ali lahko dehidrirate,
- če imate v krvi neravnovesje ionov (neravnovesje elektrolitov, ki ga lahko odkrijete pri preiskavah krvi),
- če ste imeli težave z očmi, boste morda potrebovali dodatno spremljanje oči.
- če imate hudo kožno reakcijo.
Pomanjkanje dihidropirimidin dehidrogenaze (DPD): Pomanjkanje DPD je redka bolezen, prisotna ob rojstvu, ki na splošno ni povezana z zdravstvenimi težavami, razen če jemljemo nekatera zdravila. Če imate neznano pomanjkanje DPD in jemljete zdravilo Xeloda, se lahko v hudi obliki pojavijo neželeni učinki, navedeni v poglavju 4 "Možni neželeni učinki". Povejte svojemu zdravniku, če vas kateri od neželenih učinkov skrbi ali če opazite kateri koli neželeni učinek, ki ni omenjen v tem navodilu (glejte poglavje 4 "Možni neželeni učinki").
Otroci in mladostniki
Zdravilo Xeloda ni indicirano za zdravljenje otrok in mladostnikov. Ne dajajte zdravila Xeloda otrokom in mladostnikom.
Interakcije Katera zdravila ali živila lahko spremenijo učinek zdravila Xeloda
Druga zdravila in zdravilo Xeloda
Pred začetkom zdravljenja obvestite svojega zdravnika ali farmacevta, če jemljete, ste pred kratkim jemali ali pa boste morda začeli jemati katero koli drugo zdravilo. To je bistvenega pomena, saj lahko sočasni vnos več zdravil okrepi ali zmanjša njihov učinek. Posebno pozornost je treba nameniti v primeru sočasnega vnosa:
- zdravila za protin (alopurinol),
- zdravila, ki redčijo kri (kumarin, varfarin),
- nekatera protivirusna zdravila (sorivudin in brivudin),
- zdravila za zdravljenje epileptičnih napadov ali tremorja (fenitoin),
- interferon alfa,
- radioterapijo in nekatera zdravila za zdravljenje raka (folinska kislina, oksaliplatin, bevacizumab, cisplatin, irinotekan),
- zdravila za zdravljenje pomanjkanja folne kisline.
Zdravilo Xeloda skupaj s hrano in pijačo
Zdravilo Xeloda morate vzeti v 30 minutah po koncu obroka.
Opozorila Pomembno je vedeti, da:
Nosečnost in dojenje
Pred začetkom zdravljenja obvestite svojega zdravnika, če ste noseči, sumite ali nameravate zanositi. Zdravila Xeloda ne smete jemati, če ste noseči ali sumite, da ste noseči. Med jemanjem zdravila Xeloda ne smete dojiti. Preden vzamete to zdravilo, se posvetujte z zdravnikom ali farmacevtom.
Vpliv na sposobnost upravljanja vozil in strojev
Zdravilo Xeloda lahko povzroči omotico, slabost ali utrujenost. Zato je možno, da zdravilo Xeloda vpliva na sposobnost vožnje in upravljanja s stroji.
Zdravilo Xeloda vsebuje brezvodno laktozo
Če vam je zdravnik povedal, da imate intoleranco za nekatere sladkorje, se pred jemanjem tega zdravila posvetujte z zdravnikom.
Odmerjanje, način in čas dajanja Kako uporabljati zdravilo Xeloda: odmerjanje
Vedno jemljite to zdravilo natančno tako, kot vam je povedal zdravnik ali farmacevt. Če niste prepričani, se posvetujte z zdravnikom ali farmacevtom.
Zdravilo Xeloda lahko predpiše le zdravnik, specializiran za uporabo antineoplastičnih zdravil.
Tablete Xeloda je treba pogoltniti cele z vodo v 30 minutah po koncu obroka.
Zdravnik vam bo predpisal odmerek in režim zdravljenja, ki vam ustreza. Odmerjanje zdravila Xeloda temelji na telesni površini. To se izračuna na podlagi višine in teže. Običajni odmerek za odrasle je 1250 mg / m2 telesne površine dvakrat na dan (zjutraj in zvečer). Predlagana sta dva primera: oseba, ki tehta 64 kg in z višino 1,64 m ima telesne površine 1,7 m2 in mora dvakrat na dan vzeti 4 tablete po 500 mg in 1 tableto po 150 mg. Oseba, katere teža je 80 kg in višina 1,80 m, ima telesno površino 2,00 m2 in mora vzemite 5 tablet po 500 mg dvakrat na dan.
Tablete Xeloda se običajno jemljejo 14 dni, nato pa sledi 7 -dnevni počitek (med katerim se tablete ne jemljejo). Teh 21 dni ustreza ciklu terapije.
V kombinaciji z drugimi zdravili je lahko običajni odmerek za odrasle manjši od 1250 mg / m2 telesne površine, tablete pa je treba jemati različno dolgo (na primer vsak dan brez počitka).
Zdravnik vam bo povedal, kakšen odmerek morate vzeti, kdaj ga vzeti in kako dolgo ga morate jemati.
Zdravnik vam lahko predpiše kombinacijo 150 mg in 500 mg tablet za vsako jakost.
- Tablete vzemite zjutraj in zvečer, kot vam je predpisal zdravnik.
- Tablete vzemite v 30 minutah po koncu obroka (zajtrk in večerja).
- Pomembno je, da jemljete vsa zdravila, ki vam jih je predpisal zdravnik.
Preveliko odmerjanje Kaj storiti, če ste vzeli preveč zdravila Xeloda
Če ste vzeli večji odmerek zdravila Xeloda, kot bi smeli
Če ste vzeli večji odmerek zdravila Xeloda, kot bi smeli, se pred naslednjim odmerkom čim prej posvetujte z zdravnikom.
Če ste vzeli večji odmerek zdravila Xeloda, kot bi smeli, se lahko pojavijo naslednji neželeni učinki: slabost ali bruhanje, driska, vnetje ali razjede v črevesju ali ustih, bolečine ali krvavitve iz črevesja ali želodca ali depresija kostnega mozga (zmanjšanje vrsta krvnih celic). Če opazite katerega od teh simptomov, se takoj posvetujte z zdravnikom.
Če ste pozabili vzeti zdravilo Xeloda:
Ne vzemite izpuščenega odmerka in ne podvojite naslednjega. Namesto tega nadaljujte z običajnim odmerkom in se posvetujte z zdravnikom.
Če ste prenehali jemati zdravilo Xeloda:
Prenehanje zdravljenja s kapecitabinom ne povzroča neželenih učinkov.Če prenehate jemati kapecitabin, če jemljete kumarinske antikoagulante (ki vsebujejo npr. Fenprokumon), bo zdravnik morda moral spremeniti odmerek antikoagulanta.
Če imate dodatna vprašanja o uporabi tega izdelka, se posvetujte z zdravnikom ali farmacevtom.
Neželeni učinki Kakšni so neželeni učinki zdravila Xeloda
Kot vsa zdravila ima lahko tudi to zdravilo neželene učinke, ki pa se ne pojavijo pri vseh bolnikih.
Takoj prekinite jemanje zdravila Xeloda in se obrnite na svojega zdravnika, če se pojavi kateri od naslednjih simptomov:
- Driska: če imate povečanje za 4 ali več urinov na dan v primerjavi z običajnim odvajanjem črevesja ali nočno drisko.
- Bruhanje: če bruhate več kot enkrat v 24 urah.
- Slabost: če izgubite apetit in je količina zaužite hrane na dan veliko manjša od običajne.
- Stomatitis: če imate bolečino, pordelost, oteklino ali razjede v ustih ali grlu.
- Reakcija kože rok in nog: če imate bolečine, oteklino in pordelost ali mravljinčenje v rokah in / ali nogah.
- Vročina: če imate telesno temperaturo 38 ° C ali več.
- Okužba: če imate kakršne koli znake okužbe z bakterijami ali virusi ali drugimi organizmi.
- Bolečine v prsih: če imate bolečine v središču prsnega koša, še posebej, če se pojavijo med vadbo.
- Steven-Johnsonov sindrom: če doživite boleč rdeč ali vijoličen izpuščaj, ki se razširi in mehurji in / ali druge lezije, ki se začnejo pojavljati v sluznici (npr. Usta in ustnice), še posebej, če ste bili prej občutljivi na svetlobo, okužbe dihalni sistem (npr. bronhitis) in / ali zvišana telesna temperatura.
Če se ti neželeni učinki odkrijejo zgodaj, se običajno izboljšajo v 2-3 dneh po prekinitvi zdravljenja. Če simptomi ne prenehajo, se takoj posvetujte z zdravnikom. Zdravnik vam bo morda svetoval, da nadaljujete z jemanjem manjših odmerkov.
Poleg zgoraj naštetih so pri uporabi zdravila Xeloda sami poročali o drugih zelo pogostih neželenih učinkih, ki se lahko pojavijo pri več kot 1 od 10 bolnikov:
- bolečine v trebuhu
- izpuščaj, suha ali srbeča koža
- utrujenost
- izguba apetita (anoreksija).
Ti stranski učinki lahko postanejo resni. Zato se takoj, ko opazite neželeni učinek, nemudoma obrnite na svojega zdravnika. Zdravnik vam bo naročil, naj zmanjšate odmerek in / ali začasno prekinite zdravljenje z zdravilom Xeloda. To bo pomagalo zmanjšati verjetnost, da bo stranski učinek vztrajal ali pa se bo spremenil v resen stranski učinek.
Drugi neželeni učinki so:
Pogosti neželeni učinki (pojavijo se lahko pri največ 1 od 10 bolnikov) so:
- zmanjšanje števila belih ali rdečih krvnih celic v krvi (opaženo pri testih),
- dehidracija, hujšanje,
- pomanjkanje spanja (nespečnost), depresija,
- glavobol, zaspanost, omotica, nenormalen občutek kože (odrevenelost ali mravljinčenje), sprememba okusa,
- draženje oči, povečano solzenje, rdeče oči (konjunktivitis),
- vnetje žil (tromboflebitis),
- težko dihanje, krvavitev iz nosu, kašelj, izcedek iz nosu,
- herpes ali druge okužbe s herpesom,
- okužbe pljuč ali dihal (npr. pljučnica ali bronhitis),
- črevesne krvavitve, zaprtje, bolečine v zgornjem delu trebuha, prebavne motnje, odvečni zrak, suha usta,
- kožni izpuščaj, izpadanje las (alopecija), pordelost kože, suha koža, srbenje, razbarvanje kože, izguba kože, vnetje kože, spremembe nohtov,
- bolečine v sklepih ali okončinah (okončinah), prsih ali hrbtu,
- zvišana telesna temperatura, otekanje okončin, slabo počutje,
- težave z delovanjem jeter (opažene pri preiskavah krvi) in povišanim bilirubinom v krvi (izločajo ga jetra).
Občasni neželeni učinki (pojavijo se lahko pri manj kot 1 od 100 bolnikov) so:
- okužba krvi, okužba sečil, okužba kože, okužba nosu in grla, glivične okužbe (vključno z ustno), gripa, gastroenteritis, absces,
- mehke otekline pod kožo (lipoma),
- zmanjšanje krvnih celic, vključno s trombociti, redčenje krvi (opaženo pri testih),
- alergija,
- sladkorna bolezen, znižan kalij v krvi, podhranjenost, zvišanje trigliceridov v krvi,
- stanje zmedenosti, napadi panike, depresija razpoloženja, zmanjšan libido,
- težave pri govorjenju, motnje spomina, izguba koordinacije gibov, motnje ravnotežja, omedlevica, okvara živcev (nevropatija) in težave s čutili,
- zamegljen ali dvojni vid,
- omotica, bolečine v ušesih,
- nepravilen srčni utrip in palpitacije (aritmija), bolečine v prsih in srčni infarkt (srčni napad),
- krvni strdki v globokih venah, visok ali nizek krvni tlak, zardevanje, mraz v okončinah (okončinah), vijolične lise na koži,
- krvni strdki v venah pljuč (pljučna embolija), kolaps pljuč, izguba krvi s kašljem, astma, težko dihanje pri naporu,
- obstrukcija črevesja, nabiranje tekočine v trebuhu, vnetje tankega ali debelega črevesa, želodca ali požiralnika, bolečine v spodnjem delu trebuha, nelagodje v trebuhu, zgaga (refluks hrane iz želodca), kri v blatu,
- zlatenica (porumenelost kože in oči),
- kožne razjede in mehurji, kožne reakcije na sončno svetlobo, pordelost dlani, otekanje ali bolečina obraza,
- otekanje ali togost sklepov, bolečine v kosteh, mišična oslabelost ali togost,
- nabiranje tekočine v ledvicah, povečano uriniranje ponoči, inkontinenca, kri v urinu, povečan kreatinin v krvi (znak okvare ledvic),
- nenavadne krvavitve iz nožnice,
- oteklina (edem), mrzlica in togost.
Nekateri od teh neželenih učinkov so pogosti pri uporabi kapecitabina z drugimi zdravili za zdravljenje raka. Drugi neželeni učinki, opaženi v tem kontekstu, so:
Pogosti neželeni učinki (pojavijo se lahko pri največ 1 od 10 bolnikov) so:
- znižanje natrija, magnezija in kalcija v krvi, zvišanje krvnega sladkorja,
- živčne bolečine,
- zvonjenje v ušesih (tinitus), izguba sluha,
- vnetje žil,
- kolcanje, spremenjen glas,
- bolečina ali spremenjen / nenormalen občutek v ustih, bolečina v čeljusti,
- znojenje, nočno znojenje,
- mišični krč,
- težave pri uriniranju, kri ali beljakovine v urinu,
- podplutbe ali reakcije na mestu injiciranja (ki jih povzročijo zdravila, ki se dajejo z injekcijo hkrati).
Redki neželeni učinki (pojavijo se lahko pri največ 1 od 1.000 bolnikov) so:
- zožitev ali zamašitev solznega kanala (stenoza solznega kanala),
- odpoved jeter,
- vnetje, ki vodi v disfunkcijo ali blokado izločanja žolča (holestatski hepatitis),
- posebne spremembe na elektrokardiogramu (podaljšanje intervala QT),
- nekatere vrste aritmij (vključno z ventrikularno fibrilacijo, torsades de pointes in bradikardijo),
- vnetje oči, ki povzroča bolečino in možne težave z vidom,
- vnetje kože, ki vodi do rdečih madežev in luščenja zaradi bolezni imunskega sistema.
Zelo redki neželeni učinki (pojavijo se lahko pri največ 1 od 10.000 ljudi) vključujejo:
- hude kožne reakcije, kot so izpuščaj, razjede in mehurji, ki lahko vključujejo razjede v ustih, nosu, genitalijah, rokah, stopalih in očeh (rdeče in otekle oči).
Poročanje o stranskih učinkih
Če opazite kateri koli neželeni učinek, se posvetujte z zdravnikom, farmacevtom ali medicinsko sestro.To vključuje vse možne neželene učinke, ki niso navedeni v tem navodilu. zagotovite več informacij o varnosti tega zdravila.
Potek in zadržanje
Hraniti nedosegljivo otrokom!
Ne shranjujte pri temperaturah nad 30 ° C.
Tega zdravila ne smete uporabljati po datumu izteka roka uporabnosti, ki je naveden na zunanji ovojnini in nalepki poleg oznake "EXP". Rok uporabnosti se nanaša na zadnji dan v mesecu.
Zdravila ne smete odvreči v odpadne vode ali med gospodinjske odpadke. Vprašajte farmacevta, kako odstraniti zdravila, ki jih ne uporabljate več. Tako boste zaščitili okolje.
Sestava in farmacevtska oblika
Kaj vsebuje zdravilo Xeloda
Zdravilna učinkovina je kapecitabin (150 mg za vsako filmsko obloženo tableto).
Druge pomožne snovi so:
- Jedro tablete: brezvodna laktoza, natrijeva kroskarmeloza, hipromeloza, mikrokristalna celuloza, magnezijev stearat.
- Obloga tablete: hipromeloza, titanov dioksid (E171), rumeni in rdeči železov oksid (E172), smukec.
Izgled zdravila Xeloda in vsebina pakiranja
Svetlo breskev, bikonveksna, podolgovata filmsko obložena tableta z vtisnjenim napisom "150" na eni strani in "Xeloda" na drugi strani.
Pakiranje Xeloda 150 mg filmsko obložene tablete vsebuje 60 filmsko obloženih tablet (6 pretisnih omotov po 10 tablet).
Navodilo za uporabo vira: AIFA (Italijanska agencija za zdravila). Vsebina, objavljena januarja 2016. Prisotne informacije morda niso posodobljene.
Za dostop do najnovejše različice je priporočljivo dostopati do spletnega mesta AIFA (Italijanska agencija za zdravila). Zavrnitev odgovornosti in koristne informacije.
01.0 IME ZDRAVILA
XELODA 150 mg tablete, prevlečene s filmom
02.0 KAKOVOSTNA IN KOLIČINSKA SESTAVA
Ena filmsko obložena tableta vsebuje 150 mg kapecitabina.
Pomožne snovi z znanim učinkom:
ena filmsko obložena tableta vsebuje 15,6 mg brezvodne laktoze.
Za celoten seznam pomožnih snovi glejte poglavje 6.1.
03.0 FARMACEVTSKA OBLIKA
Filmsko obložena tableta.
Xeloda 150 mg filmsko obložene tablete so svetlo breskve, bikonveksne, podolgovate oblike, z vtisnjenim napisom "150" na eni strani in "Xeloda" na drugi strani.
04.0 KLINIČNE INFORMACIJE
04.1 Terapevtske indikacije
Zdravilo Xeloda je indicirano za adjuvantno terapijo pri bolnikih, ki so bili operirani zaradi raka debelega črevesa stopnje III (Dukes C) (glejte poglavje 5.1).
Zdravilo Xeloda je indicirano za zdravljenje metastatskega kolorektalnega raka (glejte poglavje 5.1).
Zdravilo Xeloda je indicirano za zdravljenje prve stopnje napredovalega raka želodca v kombinaciji s shemo na osnovi platine (glejte poglavje 5.1).
Zdravilo Xeloda v kombinaciji z docetakselom (glejte poglavje 5.1) je indicirano za zdravljenje bolnikov z lokalno napredovalim ali metastatskim rakom dojke po neuspešni citotoksični kemoterapiji. Prejšnje zdravljenje je moralo vključevati "antraciklin. Poleg tega je zdravilo Xeloda indicirano kot monoterapija za zdravljenje bolnic z lokalno napredovalim ali metastatskim rakom dojke po odpovedi taksana in sheme kemoterapije, ki vsebuje antraciklin, ali pri katerih antraciklin ni indiciran." nadaljnjo terapijo z antraciklini.
04.2 Odmerjanje in način uporabe
Zdravilo Xeloda sme predpisati le zdravnik, ki je specializiran za uporabo antineoplastičnih zdravil, pri vseh bolnikih pa je med prvim zdravljenjem priporočljivo skrbno spremljanje.
Če pride do hude toksičnosti ali napredovanja bolezni, je treba zdravljenje prekiniti. Izračuni standardnega in zmanjšanega odmerka na podlagi telesne površine za začetne odmerke zdravila Xeloda 1250 mg / m2 in 1000 mg / m2 so podrobno opisani v tabelah 1 oziroma 2.
Odmerjanje
Priporočeno odmerjanje (glejte poglavje 5.1):
Monoterapija
Rak debelega črevesa, danke in dojke
Pri monoterapiji je priporočeni začetni odmerek kapecitabina pri adjuvantnem zdravljenju debelega črevesa, metastatskega kolorektalnega ali lokalno napredovalega ali metastatskega raka dojke 1250 mg / m2, ki ga dajemo dvakrat na dan (zjutraj in zvečer; skupaj dnevno 2500 mg / m2). 14 dni, sledi 7-dnevni počitek. Pomožno zdravljenje pri bolnikih z rakom debelega črevesa III stopnje je priporočljivo skupaj 6 mesecev.
Združena terapija
Rak debelega črevesa, danke in želodca
Pri kombiniranem zdravljenju je treba priporočeni začetni odmerek kapecitabina zmanjšati na 800 - 1000 mg / m2, če ga dajemo dvakrat na dan 14 dni, čemur sledi 7 -dnevni počitek, ali na 625 mg / m2 dvakrat na dan, če ga dajemo neprekinjeno (glejte poglavje 5.1). V kombinaciji z irinotekanom je priporočeni začetni odmerek 800 mg / m2, če ga dajemo dvakrat na dan 14 dni, čemur sledi 7-dnevni počitek v kombinaciji z irinotekanom 200 mg / m2 1. dan. Uvedba bevacizumaba v kombiniranem režimu nima učinka na začetni odmerek kapecitabina. Pri bolnikih, ki se zdravijo s kombinacijo kapecitabina in cisplatina, je treba pred dajanjem cisplatina, v skladu s povzetkom glavnih značilnosti cisplatina, uvesti premedikacijo za vzdrževanje ustrezne hidracije in antiemetično zdravljenje. Premedikacija z antiemetiki se priporoča pri bolnikih, ki se zdravijo s kombinacijo kapecitabina in oksaliplatina, v skladu s povzetkom glavnih značilnosti oksaliplatina. Pri bolnikih z rakom debelega črevesa III stopnje je priporočljivo šestmesečno adjuvantno zdravljenje.
Rak na dojki
V kombinaciji z docetakselom je priporočeni začetni odmerek kapecitabina pri zdravljenju metastatskega raka dojke 1250 mg / m2 dvakrat na dan 14 dni, čemur sledi 7-dnevni počitek v kombinaciji z docetakselom 75 mg / m2 v 1 uri intravensko infuzijo vsake 3 tedne. Pri bolnikih, ki prejemajo kombinacijo kapecitabina in docetaksela, je treba pred dajanjem docetaksela začeti premedikacijo s peroralnim kortikosteroidom, kot je deksametazon, v skladu s povzetkom glavnih značilnosti docetaksela.
Izračun odmerka zdravila Xeloda
Preglednica 1 Izračuni standardnih in zmanjšanih odmerkov kapecitabina na podlagi telesne površine, začetni odmerek 1250 mg / m2
Preglednica 2 Izračuni standardnih in zmanjšanih odmerkov kapecitabina na podlagi telesne površine, začetni odmerek 1000 mg / m2
Prilagoditev odmerka med zdravljenjem:
Splošno
Toksičnost, ki jo povzroči dajanje kapecitabina, je mogoče obvladati s simptomatskim zdravljenjem in / ali prilagoditvijo odmerka (prekinitev zdravljenja ali zmanjšanje odmerka). Ko se odmerek zmanjša, se ga nato ne sme povečati. V primeru toksičnosti, za katero po mnenju lečečega zdravnika ni verjetno, da bo postala resna ali usodna, kot so alopecija, spremenjen okus, spremembe nohtov, lahko zdravljenje nadaljujemo v istem odmerku brez zmanjšanja ali prekinitve. Bolnike, ki jemljejo kapecitabin, je treba opozoriti, da je treba v primeru zmerne ali hude toksičnosti takoj prekiniti zdravljenje. Odmerkov kapecitabina, izključenih zaradi toksičnosti, ni mogoče nadomestiti. V primeru toksičnosti je priporočljivo spremeniti odmerek:
Preglednica 3 Shema zmanjšanja odmerka kapecitabina (3-tedenski cikel ali neprekinjeno zdravljenje)
* V skladu s skupnimi merili toksičnosti (različica 1) Nacionalne skupine za klinična preskušanja kanadskega inštituta za raka (NCIC CGT) ali skupnimi terminološkimi merili za neželene dogodke (CTCAE) programa za ocenjevanje terapije proti raku, ameriški nacionalni inštitut za raka, različica 4.0 . Za sindrom roka-stopalo in hiperbilirubinemijo glejte poglavje 4.4.
Hematologija
Bolniki z izhodiščnim številom nevtrofilcev
Spremembe odmerka zaradi toksičnosti pri uporabi kapecitabina kot 3-tedenskega cikla v kombinaciji z drugimi zdravili
Kadar se kapecitabin uporablja v 3-tedenskih ciklih v kombinaciji z drugimi zdravili, je treba prilagoditi odmerek toksičnosti v skladu s preglednico 3 zgoraj za kapecitabin in v skladu z ustreznim povzetkom glavnih značilnosti zdravila za druga zdravila. .
Na začetku zdravljenja, če je za kapecitabin ali drugo zdravilo (-e) indicirano odlog zdravljenja, je treba dajanje vseh zdravil preložiti, dokler se ne pojavijo zahteve po ponovnem dajanju vseh zdravil.
Med zdravljenjem je treba zaradi tistih strupenosti, ki jih zdravnik obravnava kot nepovezane s kapecitabinom, nadaljevati z zdravljenjem s kapecitabinom in prilagoditi odmerek drugega zdravila v skladu z ustreznimi informacijami o predpisovanju.
Če naj se druga zdravila (zdravila) trajno prekinejo, se lahko zdravljenje s kapecitabinom nadaljuje, ko so izpolnjene zahteve za ponovno uvedbo kapecitabina.
Ta pristop velja za vse indikacije in vse posebne populacije bolnikov.
Spremembe odmerka zaradi toksičnosti pri uporabi kapecitabina kot stalnega zdravljenja v kombinaciji z drugimi zdravili
Spremembe odmerka glede toksičnosti pri uporabi kapecitabina kot stalnega zdravljenja v kombinaciji z drugimi zdravili je treba izvesti v skladu s preglednico 3 zgoraj za kapecitabin in v skladu z ustreznim povzetkom glavnih značilnosti zdravila za druga zdravila.
Prilagoditev odmerka pri posameznih populacijah bolnikov:
Moteno delovanje jeter
Podatkov o varnosti in učinkovitosti ni dovolj, da bi zagotovili navodila za prilagoditev odmerka pri bolnikih z okvarjenim delovanjem jeter. Podatkov o odpovedi jeter zaradi ciroze ali hepatitisa ni.
Moteno delovanje ledvic
Kapecitabin je kontraindiciran pri bolnikih s hudo ledvično insuficienco (očistek kreatinina manjši od 30 ml / min [Cockcroft in Gault] na začetku). Incidenca neželenih učinkov stopnje 3 ali 4 pri bolnikih z zmerno okvaro ledvic (očistek kreatinina 30-50 ml / min na začetku) je večja kot pri celotni populaciji. Pri začetnem odmerku 1250 mg / m2 je priporočljivo 75-odstotno zmanjšanje pri bolnikih z zmerno ledvično okvaro na začetku. Za začetni odmerek 1000 mg / m2 pri bolnikih z zmerno ledvično okvaro na začetku ni treba zmanjšati odmerka. Začetni odmerek pri bolnikih z blago okvaro ledvic (očistek kreatinina 51-80 ml / min pri Če se pri bolniku med zdravljenjem pojavi neželeni učinek stopnje 2, 3 ali 4, je treba skrbno spremljanje in "takojšnjo prekinitev zdravljenja ter naslednji odmerek prilagoditi, kot je navedeno v zgornji tabeli 3. Če izračunani očistek kreatinina med zdravljenjem pade Pri manj kot 30 ml / min je treba zdravljenje z zdravilom Xeloda prekiniti. Ta priporočila o prilagoditvi odmerka pri ledvični okvari veljajo tako za monoterapijo kot za kombinirano uporabo (glejte tudi poglavje "Starejši" spodaj).
Upokojenci
Pri uporabi samo kapecitabina ni treba prilagajati začetnega odmerka, vendar so bolniki, stari ≥ 60 let, v primerjavi z mlajšimi preiskovanci pogosteje poročali o neželenih učinkih 3. ali 4. stopnje, povezanih z zdravljenjem.
Pri uporabi kapecitabina v kombinaciji z drugimi zdravili so imeli starejši bolniki (≥ 65 let) več neželenih učinkov 3. in 4. stopnje, vključno s tistimi, ki so povzročili prekinitev zdravljenja, kot mlajši bolniki. Priporočljivo je skrbno spremljanje bolnikov, starih ≥ 60 let.
- V kombinaciji z docetakselom: Pri bolnikih, starih 60 let in več (glejte poglavje 5.1), so opazili povečano pojavnost neželenih učinkov 3. ali 4. stopnje in resnih neželenih učinkov, povezanih z zdravljenjem (glejte poglavje 5.1) .Začetni odmerek kapecitabina se je zmanjšal na 75% (950 mg / m2 dvakrat na dan) pri bolnikih, starih 60 let ali več.Če pri bolnikih, starih ≥ 60 let, zdravljenih z zmanjšanim začetnim odmerkom kapecitabina v kombinaciji z docetakselom, ne pride do toksičnosti, lahko odmerek kapecitabina previdno povečamo na 1250 mg / m2 dvakrat dnevno.
Pediatrična populacija
Kapecitabina pri pediatrični populaciji ni ustrezno uporabiti pri indikacijah raka debelega črevesa, debelega črevesa, danke in želodca.
Način dajanja
Tablete Xeloda je treba pogoltniti z vodo 30 minut po koncu obroka.
04.3 Kontraindikacije
• Zgodovina hudih ali nepričakovanih reakcij na zdravljenje s fluoropirimidinom.
• Preobčutljivost za kapecitabin ali katero koli pomožno snov, navedeno v poglavju 6.1, ali za fluorouracil.
• Pri bolnikih z znano popolno odsotnostjo aktivnosti dihidropirimidin dehidrogenaze (DPD) (glejte poglavje 4.4).
• Med nosečnostjo in dojenjem.
• Pri bolnikih s hudimi oblikami levkopenije, nevtropenije ali trombocitopenije.
• Pri bolnikih s hudo okvaro jeter.
• Pri bolnikih s hudo okvaro ledvic (očistek kreatinina manjši od 30 ml / min).
• Med zdravljenjem s sorivudinom ali njegovimi kemijsko sorodnimi analogi, kot je brivudin (glejte poglavje 4.5).
• Če obstajajo kontraindikacije za katero koli zdravilo v kombiniranem režimu, se tega zdravila ne sme uporabljati.
04.4 Posebna opozorila in ustrezni previdnostni ukrepi za uporabo
The toksičnost, ki omejuje odmerek vključujejo drisko, bolečine v trebuhu, slabost, stomatitis in sindrom roke-noge (kožna reakcija roke-noge, palmarno-plantarna eritrodisestezija). Večina neželenih učinkov je reverzibilnih in ne zahtevajo trajne prekinitve zdravljenja, čeprav je morda potrebna prekinitev ali zmanjšanje odmerka.
Driska. Bolnike s hudo drisko je treba skrbno spremljati in jim v primeru dehidracije dati tekočine in elektrolite. Lahko se predpišejo standardna zdravila proti diareji (npr. Loperamid). Driska 2. stopnje po skupnih merilih toksičnosti NCIC pomeni povečanje s 4 na 6 izpustov na dan ali nočne izpuste, pri driski 3. stopnje povečanje za 7 do 9 izpustov na dan ali inkontinenco in malabsorpcijo ter pri driski 4. stopnje an povečanje ≥ 10 izcedkov na dan ali močno krvavitev driske ali potreba po parenteralni podpori. Po potrebi je treba zmanjšati odmerek (glejte poglavje 4.2).
Dehidracija. Dehidracijo je treba preprečiti ali popraviti, ko se pojavi. Bolniki z anoreksijo, astenijo, slabostjo, bruhanjem ali drisko lahko hitro dehidrirajo. Dehidracija lahko povzroči akutno odpoved ledvic, zlasti pri bolnikih z že obstoječo ledvično okvaro ali če se kapecitabin daje v kombinaciji z znanimi nefrotoksičnimi zdravili. Akutna odpoved ledvic, ki je posledica dehidracije, je lahko potencialno usodna. Če pride do dehidracije stopnje 2 (ali višje), je treba zdravljenje s kapecitabinom nemudoma prekiniti in odpraviti dehidracijo. Po potrebi je treba odmerek prilagoditi za nastanek neželenega dogodka (glejte poglavje 4.2).
Sindrom roke in noge (znana tudi kot kožna reakcija rok in stopal ali palmarno-plantarna eritrodisestezija ali eritem okončin, ki ga povzroča kemoterapija). Sindrom roke-noge stopnje 1 je opredeljen kot odrevenelost, disestezija / parestezija, mravljinčenje, neboleč edem ali eritem rok in / ali stopal in / ali nelagodje, ki ne ovira bolnikovih običajnih dejavnosti.
Sindrom roke in noge 2. stopnje je opredeljen kot boleč eritem in edem v rokah in / ali nogah in / ali nelagodje, ki vpliva na bolnikove vsakodnevne dejavnosti.
Sindrom roke-noge stopnje 3 je opredeljen kot mokro luščenje, razjede, mehurji in hude bolečine v rokah in / ali nogah in / ali hudo nelagodje, ki bolniku onemogoča delo ali opravljanje vsakodnevnih dejavnosti. -pojavi se sindrom stopala, prekinite dajanje kapecitabina, dokler se intenzivnost simptomov ne razreši ali zmanjša na 1. stopnjo. Ko se kapecitabin in cisplatin uporabljata v kombinaciji, uporaba vitamina B6 (piridoksin) za simptomatsko ali sekundarno profilaktično zdravljenje sindroma roke-stopala ni priporočljiva, saj so objavljeni primeri pokazali, da lahko zmanjša učinkovitost cisplatina. Obstaja nekaj dokazov, da je dekspantenol učinkovit pri preprečevanju sindroma roke-noge pri bolnikih, zdravljenih z zdravilom Xeloda.
Kardiotoksičnost. Zdravljenje s fluoropirimidinom je bilo povezano s kardiotoksičnostjo, vključno z miokardnim infarktom, angino pektoris, aritmijo, kardiogenim šokom, nenadno smrtjo in elektrokardiografskimi spremembami (vključno z zelo redkimi primeri podaljšanja intervala QT). Ti neželeni učinki se lahko pogosteje pojavijo pri bolnikih s predhodno anamnezo koronarne arterije Pri bolnikih, ki so jemali kapecitabin, so poročali o srčni aritmiji (vključno z ventrikularno fibrilacijo, torsades de pointes in bradikardijo), angini pektoris, miokardnem infarktu, srčnem popuščanju in kardiomiopatiji.
Hipo- ali hiperkalcemija. Med zdravljenjem s kapecitabinom so poročali o primerih hipo- ali hiperkalciemije. Posebna previdnost je potrebna pri bolnikih z anamnezo hipo- ali hiperkalciemije v anamnezi (glejte poglavje 4.8).
Bolezni osrednjega ali perifernega živčnega sistema. Bolnike z boleznimi osrednjega ali perifernega živčnega sistema, na primer metastazami v možganih ali nevropatijo, je treba pregledati previdno (glejte poglavje 4.8).
Diabetes mellitus ali motnje elektrolitov. Bolnike s sladkorno boleznijo ali motnjami elektrolitov je treba glede na možnost poslabšanja med zdravljenjem s kapecitabinom obravnavati previdno.
Antikoagulanti, pridobljeni iz kumarina. V študiji interakcij z dajanjem enkratnega odmerka varfarina je prišlo do znatnega povečanja povprečne AUC (+ 57%) S-varfarina. Ti podatki kažejo na "interakcijo, verjetno zaradi" inhibicije izoencima 2C9 citokroma P450 s kapecitabinom. Bolnike, ki jemljejo peroralne antikoagulante, pridobljene s kumarinom, skupaj s kapecitabinom je treba redno spremljati glede možnih sprememb parametrov koagulacije (INR ali protrombina). čas) in odmerek antikoagulantov je treba ustrezno prilagoditi (glejte poglavje 4.5).
Moteno delovanje jeter. Ker ni podatkov o varnosti in učinkovitosti pri bolnikih z okvarjenim delovanjem jeter, je treba uporabo kapecitabina pri bolnikih z blago do zmerno jetrno disfunkcijo natančno spremljati, ne glede na prisotnost ali odsotnost jetrnih metastaz. Dajanje kapecitabina je treba prekiniti, če z zdravljenjem povezanih zvišanj bilirubina, večjih od 3,0 x ZMN ali z zdravljenjem povezanih zvišanj jetrnih aminotransferaz (ALT, AST), večjih od 2,5 x ZMN. x ULN.
Moteno delovanje ledvic. Incidenca neželenih učinkov stopnje 3 ali 4 pri bolnikih z zmerno okvaro ledvic (očistek kreatinina 30-50 ml / min) je večja kot pri celotni populaciji (glejte poglavji 4.2 in 4.3).
Pomanjkanje dihidropirimidin dehidrogenaze (DPD): Redka, nepričakovana in huda toksičnost (npr. Stomatitis, driska, mukozitis, nevtropenija in nevrotoksičnost), povezana s 5-FU, je bila povezana s pomanjkanjem aktivnosti DPD.
Bolniki z nizko ali brez aktivnosti DPD, encima, ki sodeluje pri razgradnji fluorouracila, imajo večje tveganje za hude, smrtno nevarne ali usodne neželene učinke, ki jih povzroča fluorouracil. Čeprav pomanjkanja DPD ni mogoče natančno identificirati, je znano, da bolniki z določenimi homozigotnimi ali sestavljenimi heterozigotnimi mutacijami genskega lokusa DPYD, ki povzročajo popolno ali skoraj popolno odsotnost encimske aktivnosti DPD (kot je bilo ugotovljeno z laboratorijsko analizo), imajo največje tveganje za smrtno nevarno ali smrtno strupenost in se jih ne sme zdraviti z zdravilom Xeloda (glejte poglavje 4.3). Noben odmerek ni bil varen za bolnike s popolno odsotnostjo aktivnosti DPD.
Bolniki z delnim pomanjkanjem DPD (na primer tisti s heterozigotnimi mutacijami v DPYD), za katere velja, da koristi zdravila Xeloda prevladajo nad njegovimi tveganji (ob upoštevanju ustreznosti alternativnega režima kemoterapije brez fluopirimidina), je treba zdraviti zelo previdno in jih pogosto spremljati s prilagajanjem odmerka glede na toksičnost. priporočajo poseben odmerek pri bolnikih z delno aktivnostjo DPD, merjeno s posebnim testom.
Pri bolnikih z neznano pomanjkljivostjo DPD, ki se zdravijo s kapecitabinom, se lahko pojavijo smrtno nevarne toksičnosti, kot so epizode akutnega prevelikega odmerjanja (glejte poglavje 4.9). V primeru akutne toksičnosti stopnje 2-4 je treba zdravljenje takoj prekiniti. O trajni prekinitvi zdravljenja je treba razmisliti na podlagi klinične ocene začetka, trajanja in resnosti ugotovljenih toksičnosti.
Oftalmološki zapleti: Bolnike je treba skrbno spremljati glede oftalmoloških zapletov, kot so keratitis in motnje roženice, še posebej, če so že imeli anamnezo očesnih motenj. Zdravljenje očesnih motenj je treba začeti na klinično ustrezen način.
Hude kožne reakcije: Xeloda lahko povzroči hude kožne reakcije, kot sta Stevens-Johnsonov sindrom in toksična epidermalna nekroliza. Pri bolnikih, pri katerih se med zdravljenjem z zdravilom Xeloda pojavi huda kožna reakcija, je treba to zdravilo trajno ukiniti.
Ker to zdravilo vsebuje pomožno snov brezvodno laktozo, tega zdravila ne smejo jemati bolniki z redkimi dednimi oblikami intolerance za galaktozo, pomanjkanjem encima Lapp laktaze in malabsorpcijo glukoze-galaktoze.
04.5 Interakcije z drugimi zdravili in druge oblike interakcij
Študije interakcij so bile izvedene samo pri odraslih.
Interakcije z drugimi zdravili:
Substrati citokroma P-450 2C9: Poleg študij varfarina med kapecitabinom in drugimi substrati CYP2C9 niso bile izvedene uradne študije medsebojnega delovanja zdravil in zdravil. Pri dajanju kapecitabina skupaj s substrati 2C9 (npr. Fenitoinom) je potrebna previdnost. Glejte tudi interakcijo z drugimi antikoagulanti, pridobljenimi iz kumarina, in poglavje 4.4.
Antikoagulanti, pridobljeni iz kumarinaPri bolnikih, ki so se sočasno zdravili s kapecitabinom in antikoagulanti, pridobljenimi iz kumarina, kot sta varfarin in fenprokumon, so poročali o spremembah parametrov strjevanja krvi in / ali krvavitvah. Te reakcije so se pojavile v obdobju nekaj dni do nekaj mesecev po začetku zdravljenja s kapecitabinom in v nekaterih primerih v enem mesecu po prekinitvi zdravljenja s kapecitabinom. V klinični študiji farmakokinetičnih interakcij je po dajanju enkratnega 20 mg odmerka varfarina zdravljenje s kapecitabinom povečalo AUC S-varfarina za 57% s povečanjem INR za 91%. Ker presnova R-varfarina ni bila spremenjena, ti podatki kažejo, da kapecitabin zmanjšuje izoencim 2C9, vendar nima učinka na izoencime 1A2 in 3A4. Bolnike, ki jemljejo antikoagulante iz kumarina sočasno s kapecitabinom, je treba redno spremljati glede možnih sprememb v parametre koagulacije (PT ali INR) in odmerek antikoagulantov je treba ustrezno prilagoditi.
Fenitoin: Med sočasno uporabo kapecitabina in fenitoina so zabeležili povečanje plazemske koncentracije fenitoina, kar je v posameznih primerih povzročilo simptome zastrupitve s fenitoinom. Bolnike, ki sočasno jemljejo fenitoin s kapecitabinom, je treba redno spremljati glede pojava povišanih plazemskih koncentracij fenitoina.
Folna kislina / folna kislina: Študija, ki je vključevala kombinacijo kapecitabina in folinske kisline, je pokazala, da folinska kislina nima pomembnega vpliva na farmakokinetiko kapecitabina in njegovih presnovkov. Vendar pa folinska kislina vpliva na farmakodinamiko kapecitabina, katerega toksičnost lahko poveča folna kislina: največji dopustni odmerek (MTD) monoterapije s kapecitabinom v intermitentnih režimih je 3000 mg / m2 na dan, medtem ko je bil kapecitabin povezan s folinsko kislino ( 30 mg dvakrat na dan) je največji dopustni odmerek padel na le 2000 mg / m2 na dan. Povečanje toksičnosti je lahko pomembno pri prehodu s 5-FU / LV na shemo na osnovi kapecitabina. .
Sorivudin in njegovi analogi: Med sorivudinom in 5-FU so poročali o klinično pomembni interakciji med zdravili, ki je posledica zaviranja sorivudina dihidropirimidin dehidrogenaze. Ta interakcija, ki vodi do povečane toksičnosti za fluoropirimidin, je lahko usodna. Zaradi tega kapecitabina ne smemo dajati sočasno s sorivudinom ali njegovimi kemijsko sorodnimi analogi, kot je brivudin (glejte poglavje 4.3). Med koncem zdravljenja s sorivudinom ali njegovimi kemijsko sorodnimi analogi, kot je brivudin, in začetkom zdravljenja s kapecitabinom je treba upoštevati vsaj 4 tedne počitka.
Antacidi: Preučevali so učinek antacida, ki vsebuje aluminijev in magnezijev hidroksid, na farmakokinetiko kapecitabina. Rahlo se je povečala plazemska koncentracija kapecitabina in njegovega presnovka (5 "-DFCR); ni vplival na 3 glavne presnovke (5 "-DFUR, 5-FU in FBAL).
Alopurinol: Opazili so medsebojno delovanje 5-FU z alopurinolom z možno zmanjšano učinkovitostjo 5-FU. Izogibati se je treba sočasni uporabi alopurinola in kapecitabina.
Interferon alfa: največji dopustni odmerek (MTD) kapecitabina je bil 2000 mg / m2 na dan, če ga jemljemo v kombinaciji z interferonom alfa-2a (3 MIU / m2 na dan), v primerjavi s 3000 mg / m2 na dan, ko smo kapecitabin dajali sami.
Radioterapija: Največji dopustni odmerek (MTD) monoterapije s kapecitabinom z uporabo intermitentnega režima je 3000 mg / m2 na dan, medtem ko je v kombinaciji z radioterapijo za rak danke največji dopustni odmerek (MTD) kapecitabina 2000 mg / m2 na dan, z uporabo bodisi neprekinjeno ali dnevno odmerjanje od ponedeljka do petka v povezavi s 6-tedenskim ciklom zdravljenja z radioterapijo.
Oksaliplatin: Pri dajanju kapecitabina v kombinaciji z oksaliplatinom ali v kombinaciji z oksaliplatinom in bevacizumabom ni bilo klinično pomembne razlike v izpostavljenosti kapecitabinu ali njegovim presnovkom, prosti platini ali celotni platini.
Bevacizumab: Klinično pomembnega učinka bevacizumaba na farmakokinetične parametre kapecitabina ali njegovih presnovkov v prisotnosti oksaliplatina ni bilo.
Interakcija s hrano
V vseh kliničnih študijah so bolnikom svetovali, naj jemljejo kapecitabin 30 minut po obroku. Ker trenutni podatki o varnosti in učinkovitosti temeljijo na dajanju zdravila s hrano, je priporočljivo, da kapecitabin jemljete s hrano, saj s hrano zmanjša hitrost absorpcije kapecitabina (glejte poglavje 5.2).
04.6 Nosečnost in dojenje
Ženske v rodni dobi / kontracepcija pri moških in ženskah
Ženskam v rodni dobi je treba svetovati, naj se med zdravljenjem s kapecitabinom izognejo tveganju nosečnosti. Če med zdravljenjem s kapecitabinom nastopi nosečnost, je treba bolnika obvestiti o možnem tveganju za plod. Med zdravljenjem je treba uporabiti učinkovito metodo kontracepcije.
Nosečnost
Študije s kapecitabinom pri nosečnicah niso bile izvedene; vendar se lahko domneva, da lahko kapecitabin, če ga dajemo nosečnicam, škoduje plodu. V študijah strupenosti za razmnoževanje pri živalih je dajanje kapecitabina povzročilo embrionalno smrtnost in teratogenost. Ti rezultati so pričakovani učinki derivatov fluoropirimidina. Kapecitabin je med nosečnostjo kontraindiciran.
Čas hranjenja
Ni znano, ali se kapecitabin izloča v materino mleko. V doječem mišjem mleku so našli pomembne količine kapecitabina in njegovih presnovkov. V obdobju zdravljenja s kapecitabinom je treba dojenje prekiniti.
Plodnost
Podatkov o zdravilu Xeloda in njegovem vplivu na plodnost ni. Ključne študije zdravila Xeloda so vključevale ženske v rodni dobi in moške le, če so bile pripravljene uporabljati ustrezno kontracepcijo, da bi se izognile nosečnosti v celotni študiji in v razumnem obdobju po njej.
V študijah na živalih so opazili učinke na plodnost (glejte poglavje 5.3).
04.7 Vpliv na sposobnost vožnje in upravljanja s stroji
Kapecitabin ima blag ali zmeren vpliv na sposobnost vožnje in upravljanja s stroji. Kapecitabin lahko povzroči omotico, utrujenost in slabost.
04.8 Neželeni učinki
Povzetek varnostnega profila
Celotni varnostni profil kapecitabina temelji na podatkih več kot 3000 bolnikov, zdravljenih samo s kapecitabinom ali s kapecitabinom v kombinaciji z različnimi režimi kemoterapije pri več indikacijah. Varnostni profili monoterapije s kapecitabinom pri populacijah bolnikov z metastatskim rakom dojke, metastatskim kolorektalnim rakom in adjuvantnim rakom debelega črevesa so podobni. Glejte poglavje 5.1 za podrobnosti o glavnih študijah, vključno z načrti študij in ključnimi rezultati učinkovitosti.
Najpogosteje poročani in / ali klinično pomembni neželeni učinki zdravil, povezani z zdravljenjem, so bili prebavne motnje (zlasti driska, slabost, bruhanje, bolečine v trebuhu, stomatitis), sindrom roke-noge (palmarno-plantarna eritrodisestezija), utrujenost, astenija, anoreksija, kardiotoksičnost, poslabšanje delovanja ledvic, kjer je bila funkcija že prej oslabljena, in tromboza / embolija.
Povzetek neželenih učinkov v obliki tabele
Neželeni učinki, za katere je preiskovalec menil, da so verjetno, verjetno ali na daljavo povezani z dajanjem kapecitabina, so navedeni v preglednici 4 za samo jemanje kapecitabina in v preglednici 5 za jemanje kapecitabina v kombinaciji z različnimi režimi kemoterapije pri več indikacijah. Za razvrščanje neželenih učinkov glede na njihovo pogostost se uporabljajo naslednji izrazi: zelo pogosti (≥ 1/10), pogosti (≥ 1/100,
Monoterapija s kapecitabinom:
V preglednici 4 so navedeni neželeni učinki, povezani z uporabo monoterapije s kapecitabinom, ki temeljijo na združeni analizi varnostnih podatkov treh glavnih študij, vključno z več kot 1900 bolniki (študije M66001, SO14695 in SO14796). Neželeni učinki so bili vključeni v posebno skupino pogostnosti glede na "splošno incidenco, ki izhaja iz skupne analize".
Preglednica 4 Povzetek povezanih neželenih učinkov, o katerih so poročali pri bolnikih, zdravljenih s monoterapijo s kapecitabinom.
Kapecitabin v kombinirani terapiji:
V preglednici 5 so navedeni neželeni učinki, povezani z uporabo kapecitabina v kombinaciji z različnimi shemami kemoterapije, pri več indikacijah, ki temeljijo na podatkih o varnosti več kot 3000 bolnikov. opazili v ključnih kliničnih študijah in le, če so dodani tistim, ki so jih opazili pri monoterapiji s kapecitabinom, ali če spadajo v skupino pogostejših kot monoterapija s kapecitabinom (glejte tabelo 4). Občasni neželeni učinki, o katerih so poročali pri kapecitabinu pri kombinirani terapiji, so v skladu z neželenimi učinki, o katerih so poročali pri monoterapiji s kapecitabinom ali pri monoterapiji s kombiniranimi zdravili (v literaturi in / ali njihovem povzetku glavnih značilnosti zdravila).
Nekateri neželeni učinki so reakcije, ki jih pogosto opazimo s kombiniranim zdravilom (npr. Periferna senzorična nevropatija z docetakselom ali oksaliplatinom, hipertenzija z bevacizumabom); poslabšanja, ki jih povzroči zdravljenje s kapecitabinom, pa ni mogoče izključiti.
Preglednica 5 Povzetek neželenih učinkov, o katerih so poročali pri bolnikih, zdravljenih s kapecitabinom v kombinirani terapiji, poleg tistih, ki so jih opazili samo s kapecitabinom ali so jih opazili v skupini z večjo pogostnostjo kot samo kapecitabin.
+ Za vsak termin je bila pogostost izračunana na podlagi ADR vseh razredov. Za izraze, označene z "+", je bila frekvenca izračunana na podlagi ADR-jev razreda 3-4. Neželeni učinki so bili vključeni glede na največjo incidenco, ugotovljeno v ključnih kliničnih preskušanjih kombinirane terapije.
Opis izbora neželenih učinkov
Sindrom roke in noge (glejte poglavje 4.4):
V študijah monoterapije s kapecitabinom (vključno s študijami adjuvantne terapije pri raku debelega črevesa, zdravljenju metastatskega kolorektalnega raka in zdravljenju raka dojke) z 1250 mg / m2 kapecitabina dvakrat na dan v dneh od 1 do 14 vsake tri tedne, sindrom roke-stopala katere koli stopnje so opazili s frekvenco od 53% do 60%; v skupini s kapecitabinom / docetakselom za zdravljenje metastatskega raka dojke je bila pogostnost 63%. Pri kombiniranem zdravljenju s kapecitabinom, s kapecitabinom 1000 mg / m2 dvakrat na dan od 1. do 14. dneva vsake tri tedne so opazili kakršno koli stopnjo sindroma roke-stopala s pogostostjo od 22% do 30%.
Kot del metaanalize 14 kliničnih preskušanj s podatki več kot 4700 bolnikov, zdravljenih z monoterapijo s kapecitabinom ali kapecitabinom v kombinaciji z različnimi shemami kemoterapije pri več indikacijah (rak debelega črevesa, kolorektalni, želodca in rak dojke), sindrom roke-noge kakršna koli stopnja se je pojavila pri 2066 bolnikih (43%) po mediani 239 dni (95% IZ: 201, 288) od začetka zdravljenja s kapecitabinom. V vseh študijah skupaj je bila ugotovljena "statistično pomembna povezava med naslednjimi kovarijatami in povečanim tveganjem za razvoj sindroma roke-stopala: povečanje začetnega odmerka kapecitabina (gram), zmanjšanje kumulativnega odmerka kapecitabina (0,1 * kg), povečanje relativnega intenzivnost odmerka v prvih 6 tednih, podaljšano trajanje študijskega zdravljenja (tedni), višja starost (10-letno povečanje), ženski spol in dobro izhodiščno stanje uspešnosti ECOG (0 v primerjavi s ≥ 1).
Driska (glejte poglavje 4.4):
Kapecitabin lahko povzroči nastanek driske, ki so jo opazili pri do 50% bolnikov.
Rezultati metaanalize 14 kliničnih študij s podatki več kot 4700 bolnikov, zdravljenih s kapecitabinom, so pokazali, da je v vseh študijah skupaj obstajala "statistično pomembna povezava med naslednjimi kovarijatami in povečanim tveganjem za nastanek driske: povečan začetni odmerek kapecitabin (gram), podaljšano trajanje študijskega zdravljenja (tedni), višja starost (10-letno povečanje) in ženski spol. Opazili so statistično pomembno povezavo med naslednjimi kovarijatami in zmanjšanjem tveganja za nastanek driske: povečanjem kumulativnega odmerka kapecitabina (0,1 * kg) in povečanjem relativne intenzivnosti odmerka v prvih 6 tednih.
Kardiotoksičnost (glejte poglavje 4.4):
Poleg neželenih učinkov, opisanih v preglednicah 4 in 5, na podlagi "združene analize podatkov o klinični varnosti iz 7 kliničnih študij, vključno z 949 bolniki (2 študiji faze III in 5 faze II pri metastatskem kolorektalnem raku) in pri metastatskem raku dojke), v povezavi z uporabo samo kapecitabina so opazili naslednje neželene učinke z incidenco manj kot 0,1%: kardiomiopatijo, srčno popuščanje, nenadno smrt in ventrikularne ekstrasistole.
Encefalopatija:
Poleg neželenih učinkov, opisanih v preglednicah 4 in 5, je bila na podlagi zgoraj omenjene združene analize podatkov o klinični varnosti iz 7 kliničnih študij uporaba samo kapecitabina povezana tudi z encefalopatijo, pri kateri je incidenca manjša od 0,1%.
Posebne populacije
Starejši bolniki (glejte poglavje 4.2):
"Analiza varnostnih podatkov pri bolnikih, starih ≥ 60 let, zdravljenih z monoterapijo s kapecitabinom, in" analiza bolnikov, zdravljenih s terapevtsko kombinacijo kapecitabina in docetaksela, je pokazala povečano pojavnost neželenih učinkov stopnje 3 in 4, povezanih z zdravljenjem in z zdravljenjem resni neželeni učinki v primerjavi z bolniki, mlajšimi od 60 let. Poleg tega so bolniki, stari ≥ 60 let, zdravljeni s kapecitabinom in docetakselom, predčasno prekinili zdravljenje zaradi pogostejših neželenih učinkov kot bolniki, mlajši od 60 let.
Rezultati metaanalize 14 kliničnih študij s podatki več kot 4700 bolnikov, zdravljenih s kapecitabinom, so pokazali, da je v vseh študijah skupaj obstajala "statistično pomembna povezava med" napredovanjem starosti (10-letno povečanje) in povečanim tveganjem. razvoj sindroma roke-noge in driske ter zmanjšano tveganje za nastanek nevtropenije.
Seks
Rezultati metaanalize 14 kliničnih preskušanj s podatki več kot 4700 bolnikov, zdravljenih s kapecitabinom, so pokazali, da je v vseh študijah skupaj "statistično pomembna povezava med ženskim spolom in povečanim tveganjem za razvoj sindroma. drisko in zmanjšano tveganje za nastanek nevtropenije.
Bolniki z okvaro ledvic (glejte poglavja 4.2, 4.4 in 5.2):
Analiza podatkov o varnosti pri bolnikih, zdravljenih z monoterapijo s kapecitabinom (kolorektalni rak) z okvaro ledvic na začetku, je pokazala povečano pojavnost neželenih učinkov, povezanih z zdravljenjem 3 in 4 stopnje, v primerjavi z bolniki z normalno ledvično okvaro (36% pri bolnikih brez okvare ledvic n = 268 proti 41% pri blagi okvari n = 257 oziroma 54% pri zmerni n = 59) (glejte poglavje 5.2). Pri bolnikih z zmerno okvarjenim delovanjem ledvic so opazili povečanje stopnje zmanjšanja odmerka (44%) v primerjavi s 33% in 32% pri bolnikih z blago ali brez ledvične okvare ter povečanje prezgodnje prekinitve zdravljenja (21% prekinitev v prvih dveh ciklov) v primerjavi s 5% in 8% pri bolnikih z malo ali brez ledvične okvare.
Poročanje o domnevnih neželenih učinkih
Poročanje o domnevnih neželenih učinkih, ki se pojavijo po registraciji zdravila, je pomembno, saj omogoča stalno spremljanje razmerja med koristmi in tveganji zdravila. Zdravstvene delavce prosimo, da o vsakem sumu na neželene učinke poročajo prek nacionalnega sistema za poročanje www .agenziafarmaco.gov .it / it / odgovorno.
04.9 Preveliko odmerjanje
Manifestacije akutnega prevelikega odmerjanja vključujejo slabost, bruhanje, drisko, mukozitis, draženje prebavil in krvavitve ter depresijo kostnega mozga. Klinično obvladovanje prevelikega odmerjanja je treba izvesti s konvencionalno terapijo in podporno medicinsko intervencijo, da se odpravijo prisotne klinične manifestacije in preprečijo morebitni zapleti.
05.0 FARMAKOLOŠKE LASTNOSTI
05.1 Farmakodinamične lastnosti
Farmakoterapevtska skupina: citostatik (antimetabolit).
Oznaka ATC: L01BC06.
Kapecitabin je necitotoksičen fluoropirimidin karbamat, ki deluje kot peroralni predhodnik citotoksične oblike 5-fluorouracil (5-FU). Kapecitabin se aktivira z več encimskimi koraki (glejte poglavje 5.2). Encim, ki sodeluje pri končni pretvorbi v 5-FU, timidin fosforilazo (ThyPase), najdemo v tumorskih tkivih, pa tudi v normalnih tkivih, čeprav na splošno v nižji koncentraciji. sinergijski učinek v kombinaciji z docetakselom, ki je lahko povezan s hiperegulacijo timidin fosforilaze z docetakselom.
Ugotovljeno je bilo, da presnova 5-FU na anabolični poti blokira reakcijo metilacije deoksiuridilne kisline v timidilno kislino in tako posega v sintezo deoksiribonukleinske kisline (DNA). Vključitev 5-FU vodi tudi v zaviranje sinteze RNA in beljakovin. Ker sta DNA in RNA bistveni za delitev in rast celic, lahko 5-FU povzroči pomanjkanje timidina, kar povzroči neuravnoteženo rast in celično smrt. Učinki pomanjkanja DNA in RNA so še posebej izraziti v celicah, ki rastejo hitreje in se hitreje presnavljajo 5-FU.
Rak debelega črevesa in danke:
Monoterapija s kapecitabinom pri adjuvantnem zdravljenju raka debelega črevesa
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III pri bolnikih z rakom debelega črevesa stopnje III (Dukes C) podpirajo uporabo kapecitabina za adjuvantno terapijo pri bolnikih z rakom debelega črevesa (študija X-ACT, M66001). bolniki so bili naključno izbrani za zdravljenje s kapecitabinom (1250 mg / m2 dvakrat na dan 2 tedna, čemur je sledil 1 teden počitka, kot 3-tedenski cikli za 24 tednov) ali 5-FU in levkovorin (urnik klinike Mayo: 20 mg / m2 IV levkovorin sledi 425 mg / m2 IV 5-FU bolus, 1. do 5. dan, vsakih 28 dni 24 tednov) .Kapecitabin je bil vsaj enakovreden 5-FU / LV IV pri preživetju brez bolezni pri populaciji po protokolu (HR 0,92; 95% IZ: 0,80–1,06) .Preživetje brez bolezni in skupno preživetje je pokazalo HR 0,88 (95% IZ: 0,77-1,01; p = 0,068) oziroma 0,86 (95% IZ: 0,74-1,01; p = 0,060). Mediana spremljanja v času analize je bila 6,9 leta, v predhodno načrtovani multivariatni analizi Coxa pa je bila dokazana superiornost kapecitabina nad bolusom 5-FU / LV. Za vključitev v model so bili v statistični analizi vnaprej določeni naslednji dejavniki: starost, čas od operacije do randomizacije, spol, izhodiščne ravni CEA, izhodiščne bezgavke in država. V randomizirani populaciji se je pokazalo, da je kapecitabin boljši od 5-FU / LV tako glede preživetja brez bolezni (HR: 0,849; 95% IZ: 0,739-0,976; p = 0,0212) kot glede na celotno preživetje (HR : 0,828; 95% IZ: 0,705-0,971; p = 0,0203).
Kombinirano zdravljenje pri adjuvantnem zdravljenju raka debelega črevesa
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III pri bolnikih z rakom debelega črevesa stopnje III (Dukes C) podpirajo uporabo kapecitabina v kombinaciji z oksaliplatinom (XELOX) za adjuvantno zdravljenje pri bolnikih z rakom debelega črevesa (študija NO16968).V tej študiji je bilo 944 bolnikov naključno izbranih za zdravljenje s kapecitabinom (1000 mg / m2 dvakrat na dan 2 tedna, čemur je sledil 1 teden počitka, kot 3-tedenski tečaji 24 tednov) v kombinaciji z oksaliplatinom (130 mg / m2 z intravensko infuzijo 2 uri 1. dan vsake 3 tedne); 942 bolnikov je bilo naključno izbranih za bolus 5-FU in levkovorin. V primarni analizi za DFS v populaciji ITT se je pokazalo, da je XELOX bistveno boljši od 5-FU / LV (HR = 0,80, 95% IZ = [0,69; 0,93]; p = 0, 0045). Stopnja DFS je bila v skupini XELOX 71% v primerjavi s 67% v skupini s 5-FU / LV. Analiza, opravljena za sekundarno končno točko RFS, podpira te rezultate s HR 0,78 (95% IZ = [0,67; 0,92]; p = 0,0024) v skupini XELOX v primerjavi s krakom 5-FU / LV. XELOX je pokazal trend superiornosti v smislu OS s HR 0,87 (95% IZ = [0,72; 1,05]; p = 0,1486), kar pomeni v 13-odstotno zmanjšanje tveganja smrti.5-letni OS je bil 78% za XELOX v primerjavi s 74% za 5-FU / LV. Podatki o učinkovitosti temeljijo na povprečnem času opazovanja 59 mesecev za OS in 57 mesecev za DFS. Stopnja umika študije zaradi neželenih učinkov je bila v skupini XELOX (21%) višja kot v skupini z monoterapijo 5-FU / LV (9%) v populaciji ITT.
Monoterapija s kapecitabinom pri metastatskem kolorektalnem raku
Podatki iz dveh podobno zasnovanih, multicentričnih, randomiziranih, kontroliranih kliničnih preskušanj faze III (SO14695: SO14796) podpirajo uporabo kapecitabina za prvo linijo zdravljenja metastatskega kolorektalnega raka. / m2 dvakrat na dan 2 tedna, čemur je sledil enotedenski počitek in ga dajali v 3-tedenskih ciklusih. mg / m2 intravenski bolus 5-FU, od 1. do 5. dneva, vsakih 28 dni). raziskovalec) je bil: 25,7% (kapecitabin) v primerjavi s 16,7% (shema Mayo); str
Kombinirano zdravljenje pri prvi liniji zdravljenja metastatskega kolorektalnega raka
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III (NO16966) podpirajo uporabo kapecitabina v kombinaciji z oksaliplatinom ali v kombinaciji z oksaliplatinom in bevacizumabom za prvo linijo zdravljenja metastatskega kolorektalnega raka. Študija je vključevala dva dela: prva dva -ročni del, v katerem je bilo 634 bolnikov randomiziranih na dva različna režima zdravljenja, to je XELOX ali FOLFOX-4, in naslednji faktorjski del 2x2, v katerem je bilo 1401 bolnikov randomiziranih na štiri različne režime zdravljenja. plus placebo, XELOX plus bevacizumab in FOLFOX-4 plus bevacizumab Za sheme zdravljenja glejte tabelo 6.
Preglednica 6 Režimi zdravljenja v študiji NO16966 (mCRC)
V splošni primerjavi je bila dokazana neinferiornost rok, ki vsebujejo XELOX, v primerjavi s tistimi, ki so vsebovale FOLFOX-4 v smislu preživetja brez napredovanja bolezni pri upravičeni populaciji bolnikov in populaciji, namenjeni zdravljenju (glej tabelo 7). Rezultati kažejo, da je XELOX glede na celotno preživetje enakovreden FOLFOX-4 (glej tabelo 7). Primerjava XELOX plus bevacizumaba in FOLFOX-4 plus bevacizumaba je bila sestavljena iz "vnaprej načrtovane raziskovalne analize. Pri primerjavi teh podskupin zdravljenja je bil XELOX in bevacizumab podoben FOLFOX-4 plus bevacizumabu glede na preživetje brez napredovanja bolezni (razmerje nevarnosti 1,01; 97,5% IZ: 0,84-1,22) .Medijalno spremljanje v času primarnih analiz pri populaciji, namenjeni zdravljenju, je bilo 1,5 leta; v preglednico 7 so vključeni tudi podatki, izvedeni po nadaljnjem letu spremljanja Analiza PFS med zdravljenjem vendarle ni potrdila rezultatov analize splošnih PFS in OS: razmerje nevarnosti XELOX v primerjavi s FOLFOX -4 je bilo 1,24 z 97,5% IZ: 1,07 - 1,44. Čeprav analize občutljivosti kažejo, da razlike pri načrtovanju režima in oceni tumorja čas vpliva na tekočo analizo zdravljenja PFS, dokončne razlage za to niso našli rezultat.
Tabela 7 Ključni rezultati učinkovitosti za analizo neinferiornosti študije št. 16966
* OZO = populacija upravičenih bolnikov; ** ITT = populacija, namenjena zdravljenju.
V randomizirani, kontrolirani študiji faze III (CAIRO) so preučevali učinek uporabe kapecitabina v začetnem odmerku 1000 mg / m2 2 tedna vsake 3 tedne v kombinaciji z irinotekanom za zdravljenje prve izbire pri bolnikih z metastatsko kolorektalno. raka. 820 bolnikov je bilo naključno izbranih za zaporedno (n = 410) ali kombinirano (n = 410) zdravljenje. Zaporedno zdravljenje je obsegalo zdravljenje prve vrste s kapecitabinom (1250 mg / m2 dvakrat na dan 14 dni), drugo linijo z irinotekanom (350 mg / m2 1. dan) in tretjo linijo s kombinacijo kapecitabina. (1000 mg / mg) m2 dvakrat na dan 14 dni) in oksaliplatina (130 mg / m2 1. dan) .Kombinirano zdravljenje je obsegalo zdravljenje s kapecitabinom prve linije (1000 mg / m2 dvakrat na dan 14 dni) v kombinaciji z irinotekanom (250 mg / m2 1. dan). ) (XELIRI) in drugo linijo s kapecitabinom (1000 mg / m2 dvakrat na dan 14 dni) in oksaliplatinom (130 mg / m2 1. dan). -prosto preživetje pri populaciji, ki se namerava zdraviti, je bilo pri monoterapiji s kapecitabinom 5,8 meseca (95% IZ; 5,1 -6,2 meseca) in pri zdravilu XELIRI 7,8 meseca (95% IZ: 7,0 -8,3 meseca; p = 0,0002). to je bilo povezano s povečano incidenco gastrointestinalne toksičnosti in nevtropenije med zdravljenjem s prvo linijo zdravljenja z zdravilom XELIRI (26% oziroma 11% za zdravilo XELIRI oziroma kapecitabin prve linije).
V treh randomiziranih študijah pri bolnikih z metastatskim kolorektalnim rakom so režim XELIRI primerjali s 5-FU + irinotekanom (FOLFIRI). Režimi XELIRI so vključevali kapecitabin 1000 mg / m2 dvakrat na dan od 1. do 14. dneva v treh tednih v kombinaciji z irinotekanom 250 mg / m2 1. dan. V večji študiji (BICC-C) so bili bolniki randomizirani na odprto zdravljenje z zdravilom FOLFIRI (n = 144), bolusom 5-FU (mIFL) (n = 145) ali XELIRI (n = 141) in nadalje randomizirano na dvojno slepo celekoksib ali placebo. Mediana PFS je bila 7,6 meseca za FOLFIRI, 5,9 meseca za mIFL (p = 0,004 za primerjavo s FOLFIRI) in 5,8 meseca za XELIRI (p = 0,015). Mediana OS je bila 23,1 meseca za zdravilo FOLFIRI, 17,6 meseca za mIFL (p = 0,09) in 18,9 meseca za zdravilo XELIRI (p = 0,27). XELIRI oziroma FOLFIRI).
V študiji EORTC so bili bolniki randomizirani na odprto zdravljenje z zdravilom FOLFIRI (n = 41) ali XELIRI (n = 44), nato pa v dvojno slepi celekoksib ali placebo. Mediana PFS in splošno preživetje (OS) sta bila pri zdravilu XELIRI nižja v primerjavi s FOLFIRI (PFS 5,9 proti 9,6 meseca in OS 14,8 proti 19,9 meseca); poleg tega so pri bolnikih, ki so prejemali režim XELIRI (41% XELIRI; 5,1% FOLFIRI), poročali o prekomerni stopnji driske.
V študiji, ki jo je objavil Skof et al., so bili bolniki naključno izbrani za prejemanje FOLFIRI ali XELIRI. Skupna stopnja odziva je bila v skupini XELIRI 49% in v skupini FOLFIRI 48% (p = 0,76). Ob koncu zdravljenja 37% bolnikov v skupini XELIRI in 26% bolnikov v skupini FOLFIRI ni imelo nobenih znakov bolezni (p = 0,56). Toksičnost je bila med zdravljenji podobna, razen nevtropenije, o kateri so najpogosteje poročali pri bolnikih, zdravljenih z zdravilom FOLFIRI.
Montagnani et al. uporabili so rezultate treh zgoraj navedenih študij, da bi zagotovili "globalno analizo randomiziranih preskušanj, v kateri so primerjali terapevtske sheme FOLFIRI in XELIRI pri zdravljenju mCRC." Znatno zmanjšanje tveganja za napredovanje bolezni je bilo povezano z zdravljenjem z zdravilom FOLFIRI (HR 0,76; 95% IZ: 0,62-0,95; p
Podatki iz randomiziranega kliničnega preskušanja (Souglakos et al., 2012) primerjave med FOLFIRI + bevacizumabom in XELIRI + bevacizumabom niso pokazale pomembnih razlik med PFS in OS med zdravljenji. Bolnike smo randomizirali na zdravljenje z zdravilom FOLFIRI plus bevacizumabom (roka A, n = 167) ali XELIRI plus bevacizumabom (roka B, n = 166). Za roko B je režim XELIRI uporabljal kapecitabin 1000 mg / m2 dvakrat na dan 14 dni + irinotekan 250 mg / m2 1. dan. Za zdravljenje z zdravilom FOLFIRI-Bev in zdravljenje z zdravilom XELIRI-Bev je bilo povprečno preživetje brez napredovanja ( PFS), splošno preživetje in odziv so bili naslednji: 10,0 mesecev in 8,9 meseca (p = 0,64); 25,7 mesecev in 27,5 mesecev (p = 0,55); 45,5% in 39,8% (p = 0,32). Bolniki, zdravljeni z zdravilom XELIRI + bevacizumab, so poročali o bistveno večji pojavnosti driske, vročinske nevtropenije in kožnih reakcij na rokah in nogah v primerjavi z bolniki, zdravljenimi z zdravilom FOLFIRI + bevacizumab z znatno povečanimi zamudami zdravljenja, zmanjšanjem odmerkov in prekinitvami zdravljenja.
Podatki iz multicentrične, randomizirane, kontrolirane študije faze II (AIO KRK 0604) podpirajo uporabo kapecitabina v začetnem odmerku 800 mg / m2 2 tedna vsake 3 tedne v kombinaciji z irinotekanom in bevacizumabom za zdravljenje. bolniki z metastatskim kolorektalnim rakom.
120 bolnikov je bilo naključno izbranih na spremenjeno shemo XELIRI s kapecitabinom 800 mg / m2 dvakrat na dan dva tedna, čemur sledi 7 dni počitka), irinotekanom (200 mg / m2 v obliki 30-minutne infuzije 1. dan vsake 3 tedne) in bevacizumabom (7,5 mg / kg infundiranega 30 do 90 minut 1. dan vsake 3 tedne); 127 bolnikov je bilo naključno izbranih za zdravljenje s kapecitabinom (1000 mg / m2 dvakrat na dan dva tedna, čemur sledi 7 dni počitka), oksaliplatinom (130 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne) in bevacizumabom (7,5 mg / kg infundiranega 30 do 90 minut 1. dan vsake 3 tedne). Po povprečnem trajanju spremljanja za študijsko populacijo 26,2 meseca so bili odzivi na zdravljenje naslednji:
Tabela 8 Rezultati učinkovitosti za študijo AIO KRK
Kombinirano zdravljenje v drugi liniji zdravljenja metastatskega kolorektalnega raka
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III (NO16967) podpirajo uporabo kapecitabina v kombinaciji z oksaliplatinom za drugo linijo zdravljenja metastatskega kolorektalnega raka. z irinotekanom v kombinaciji s shemo, ki temelji na fluoropirimidinu, kot zdravljenje prve linije so bili randomizirani na zdravljenje z XELOX ali FOLFOX-4. Za režim odmerjanja XELOX in FOLFOX-4 (brez dodatka placeba ali bevacizumaba) glejte tabelo 6. Pokazalo se je, da XELOX ni slabši od FOLFOX-4 v smislu preživetja brez napredovanja bolezni v protokolu in namena zdravljenja (glej tabelo 9.) Rezultati kažejo, da je XELOX glede na celotno preživetje enakovreden FOLFOX-4 (glej tabelo 9). Mediano spremljanje v času primarnih analiz pri populaciji z namenom zdravljenja je bilo pri 2,1 letih; podatki iz analiz, opravljenih po nadaljnjih 6 mesecih spremljanja, so prav tako vključeni v tabelo 9.
Preglednica 9 Ključni rezultati učinkovitosti za analizo neinferiornosti študije NO16967
* PPP = populacija po protokolu; ** ITT = populacija, namenjena zdravljenju.
Napredni rak želodca:
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III pri bolnikih z napredovalim rakom želodca podpirajo uporabo kapecitabina pri prvi liniji zdravljenja napredovalega raka želodca (ML17032). V tej študiji je bilo 160 bolnikov randomiziranih. 1000 mg / m2 dvakrat na dan 2 tedna, čemur sledi 7 dni počitka) in cisplatin (80 mg / m2 v obliki 2-urne infuzije vsake 3 tedne) .Skupaj je bilo 156 bolnikov naključno izbranih za zdravljenje s 5-FU (800 mg / m2 na dan kot neprekinjena infuzija od 1. do 5. dne vsake 3 tedne) in cisplatin (80 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne). do 5-FU v kombinaciji s cisplatinom glede preživetja brez napredovanja bolezni v analizi po protokolu (HR 0,81; 95% IZ: 0,63-1,04). Mediano preživetje brez napredovanja je bilo 5,6 meseca (kapecitabin + cisplatin) v primerjavi s 5,0 meseca (5-FU + cisplatin). Razmerje nevarnosti za trajanje preživetja (skupno preživetje) je bilo podobno razmerju nevarnosti za preživetje brez napredovanja bolezni (HR 0,85; 95% IZ: 0,64 - 1,13). Mediano trajanje preživetja je bilo 10,5 mesecev (kapecitabin + cisplatin) v primerjavi z 9,3 meseca (5-FU + cisplatin).
Podatki iz multicentričnega, randomiziranega, kliničnega preskušanja faze III, v katerem so primerjali kapecitabin s 5-FU ter oksaliplatinom in cisplatinom pri bolnikih z napredovalim rakom želodca, podpirajo uporabo kapecitabina pri zdravljenju napredovalega raka želodca prve stopnje (REAL-2). V študiji je bilo 1002 bolnikov randomiziranih s faktorskim načrtom 2x2 v eno od naslednjih štirih vej:
-ECF: epirubicin (50 mg / m2 kot bolus 1. dan vsake 3 tedne), cisplatin (60 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne) in 5-FU (200 mg / m2 dnevno) kot se infuzija nadaljuje skozi osrednji kateter).
- ECX: epirubicin (50 mg / m2 kot bolus 1. dan vsake 3 tedne), cisplatin (60 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne) in kapecitabin (625 mg / m2 dvakrat na dan kot zdravljenje) neprekinjeno).
-EOF: epirubicin (50 mg / m2 kot bolus 1. dan vsake 3 tedne), oksaliplatin (130 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne) in 5-FU (200 mg / m2 dnevno) kot se infuzija nadaljuje skozi osrednji kateter).
- EOX: epirubicin (50 mg / m2 kot bolus 1. dan vsake 3 tedne), oksaliplatin (130 mg / m2 kot 2-urna infuzija 1. dan vsake 3 tedne) in kapecitabin (625 mg / m2 dvakrat na dan kot zdravljenje) neprekinjeno).
Primarne analize učinkovitosti v populaciji po protokolu so pokazale neinferiornost v celotnem preživetju za sheme, ki vsebujejo kapecitabin, v primerjavi s shemami na osnovi 5-FU (HR 0,86; 95% IZ: 0,8-0,0, 99) in za primerjave režimov, ki vsebujejo oksaliplatin na sheme na osnovi cisplatina (HR 0,92; 95% IZ: 0,80 - 1,1). Mediana celotnega preživetja je bila 10,9 meseca v režimih na osnovi kapecitabina in 9,6 mesecev pri tistih, ki so vsebovali 5-FU. Mediana celotnega preživetja je bila 10,0 mesecev v režimih na osnovi cisplatina in 10,4 meseca v režimih na osnovi oksaliplatina.
Kapecitabin so uporabljali tudi v kombinaciji z oksaliplatinom pri zdravljenju napredovalega raka želodca. Študije z monoterapijo s kapecitabinom kažejo, da kapecitabin kaže aktivnost pri napredovalem raku želodca.
Napredni rak želodca, debelega črevesa in debelega črevesa in danke: metaanaliza
Metaanaliza šestih kliničnih študij (študije SO14695, SO14796, M66001, NO16966, NO16967, M17032) podpira uporabo kapecitabina kot nadomestka samo za 5-FU in pri kombiniranem zdravljenju raka prebavil. Združena analiza vključuje 3097 bolnikov zdravljenih z režimi, ki vsebujejo kapecitabin, in 3074 bolnikov, zdravljenih s shemami, ki vsebujejo 5-FU. Mediana celotnega preživetja je bila 703 dni (95% IZ: 671; 745) pri bolnikih, zdravljenih s shemami, ki vsebujejo kapecitabin, in 683 dni (95% IZ: 646; 715) pri tistih, ki so se zdravili s shemami, ki vsebujejo 5-FU. Razmerje nevarnosti za celotno preživetje je bilo 0,94 (95% IZ: 0,89; 1,00, p = 0,0489), kar kaže, da režimi, ki vsebujejo kapecitabin, niso slabši od tistih, ki vsebujejo 5- FU.
Rak na dojki
Kombinirano zdravljenje s kapecitabinom in docetakselom pri lokalno napredovalem ali metastatskem raku dojke
Podatki iz multicentričnega, randomiziranega, kontroliranega kliničnega preskušanja faze III podpirajo uporabo kapecitabina v kombinaciji z docetakselom za zdravljenje bolnikov z napredujočim lokalno napredovalim ali metastatskim rakom dojke po neuspehu citotoksične kemoterapije, ki je vključevala "antraciklin". V tej študiji je bilo 255 bolnikov naključno izbranih za zdravljenje s kapecitabinom (1250 mg / m2 dvakrat na dan 2 tedna, čemur je sledil enotedenski počitek in docetaksel 75 mg / m2 kot 1-urna intravenska infuzija vsake 3 tedne). 256 bolnikov je bilo naključno izbranih za zdravljenje samo z docetakselom (100 mg / m2 kot 1 -urna intravenska infuzija vsake 3 tedne). Preživetje je bilo v skupini kombinacije kapecitabin + docetaksel boljše (p = 0,0126). Mediano preživetje je bilo 442 dni (kapecitabin + docetaksel) v primerjavi s 352 dnevi (samo docetaksel). Skupne stopnje objektivnega odziva v celotni randomizirani populaciji (ocena raziskovalca) so bile: 41,6% (kapecitabin + docetaksel) v primerjavi z 29,7% (samo docetaksel); p = 0,0058. Čas do napredovanja bolezni je bil v skupini s kapecitabinom in docetakselom boljši ( str
Monoterapija s kapecitabinom po neuspehu taksana in kemoterapije, ki vsebuje antracikline, in kjer zdravljenje z antraciklini ni indicirano
Podatki iz dveh multicentričnih kliničnih preskušanj druge faze podpirajo uporabo monoterapije s kapecitabinom za zdravljenje bolnikov, ki napredujejo po neuspešni kemoterapiji, ki je vključevala taksane in antraciklin, ali za katere ni indicirano dodatno zdravljenje. Antraciklini. od 236 bolnikov je bilo zdravljenih s kapecitabinom (1250 mg / m2 dvakrat na dan 2 tedna, čemur je sledil enotedenski počitek) .Splošna stopnja objektivnega odziva (ocena raziskovalca) je bila 20 % (prva študija) in 25 % (druga študija) Mediana časa do napredovanje je bilo 93 in 98 dni. Mediana preživetja je bila 384 in 373 dni.
Vse indikacije:
Metaanaliza 14 kliničnih preskušanj s podatki o več kot 4700 bolnikih, zdravljenih samo s kapecitabinom ali v kombinaciji z različnimi shemami kemoterapije pri več indikacijah (rak debelega črevesa, kolorektalni, želodca in dojke) je pokazala, da je splošno preživetje pri bolnikih, zdravljenih s kapecitabinom, daljše. razvil sindrom roke-noge kot pri bolnikih, ki niso: mediana celotnega preživetja 1100 dni (95% IZ: 1007, 1200) v primerjavi s 691 dnevi (95% IZ: 638; 754) z razmerjem nevarnosti 0,61 (95% IZ: 0,56 , 0,66).
Pediatrična populacija:
Evropska agencija za zdravila se je odpovedala obveznosti izvajanja študij z zdravilom Xeloda v vseh podrazredih pediatrične populacije pri adenokarcinomu debelega črevesa in danke, adenokarcinomu želodca in raku dojke (za informacije o "pediatrični uporabi" glejte poglavje 4.2).
05.2 Farmakokinetične lastnosti
Farmakokinetiko kapecitabina so ocenjevali v razponu odmerkov 502-3514 mg / m2 / dan. Parametri kapecitabina, 5 "-deoksi-5-fluorocitidina (5" -DFCR) in 5 "-deoksi-5-fluorouridina (5" DFUR), izmerjeni na 1. in 14. dan, so bili podobni. AUC 5-FU na 14. dan je bila 30-35% višja. Zmanjšanje odmerka kapecitabina zmanjšuje sistemsko izpostavljenost 5-FU bolj sorazmerno z odmerkom zaradi nelinearne farmakokinetike aktivnega presnovka.
Absorpcija
Po peroralni uporabi se kapecitabin popolnoma in hitro absorbira; nato se popolnoma pretvori v presnovke 5 "-DFCR in 5" -DFUR. Dajanje s hrano zmanjša hitrost absorpcije kapecitabina, vendar povzroči le majhen učinek na AUC 5 "-DFUR in AUC naslednjega presnovka 5-FU. V odmerku 1250 mg / m2 14. dan po obroku, največje plazemske koncentracije (Cmax v mcg / ml) kapecitabina, 5 "-DFCR, 5" -DFUR, 5 -FU in FBAL so bile 4,67 - 3,05 - 12,1 - 0,95 oziroma 5,46. Čas za dosego najvišjih plazemskih koncentracij (Tmax v urah) je bilo 1,50 - 2,00 - 2,00 - 2,00 in 3,34 Vrednosti AUC0- ∞ v mcg • h / ml so bile 7,75 - 7,24 - 24,6 - 2,03 in 36,3.
Distribucija
Opravljene študije človeške plazme in vitro je pokazala, da so kapecitabin, 5 "DFCR, 5" -DFUR in 5 -FU vezani na beljakovine, predvsem albumin, v odstotkih 54%, 10%, 62% in 10%.
Biotransformacija
Kapecitabin se najprej presnovi z jetrno karboksilestrazo v 5 "-DFCR, ki se nato s citidin deaminazo, ki se večinoma nahaja v jetrnih in tumorskih tkivih, pretvori v 5" -DFUR. Nato sledi "nadaljnja katalitična aktivacija 5" -DFUR s timidin fosforilazo (ThyPase). Encimi, vključeni v katalitično aktivacijo, so prisotni v tumorskih tkivih, pa tudi v zdravih tkivih, čeprav na splošno v manjših količinah.Zaporedna encimska biotransformacija kapecitabina v 5-FU vodi do višjih koncentracij v neoplastičnih tkivih. Zdi se, da je pri kolorektalnem raku generacija 5-FU v veliki meri lokalizirana v tumorskih stromalnih celicah. Po peroralni uporabi kapecitabina pri bolnikih s kolorektalnim rakom je bilo razmerje koncentracije 5-FU pri raku debelega črevesa in danke do sosednjih tkiv 3,2 (od 0,9 do 8,0). Razmerje koncentracije 5-FU v tumorju in plazmi je bilo 21,4 (v razponu od 3,9 do 59,9, n = 8), medtem ko je bilo razmerje v zdravem tkivu v plazmi 8,9 (z variacijo od 3,0 do 25,8, n = 8). Aktivnost timidin fosforilaze je bila izmerjena in je bila pri primarnem kolorektalnem raku 4 -krat večja od prijavljenih vrednosti v sosednjem normalnem tkivu. Na podlagi imunohistokemičnih študij se zdi, da je timidin fosforilaza v veliki meri lokalizirana v tumorskih stromalnih celicah.
5-FU se nato encim dihidropirimidin dehidrogenaza (DPD) katabolizira v dihidro-5-fluorouracil (FUH2), ki je veliko manj strupen.Dihidropirimidaza deluje na pirimidinski obroč, da pridobi 5-fluoro-ureidopropionsko kislino (FUPA). -ureido-propionaza pretvori FUPA v a-fluoro-b-alanin (FBAL), ki se izloči z urinom.Dejavnost dihidropirimidin dehidrogenaze (DPD) je kritični omejevalni dejavnik. Pomanjkanje DPD lahko povzroči "povečano toksičnost kapecitabina (glejte poglavji 4.3 in 4.4).
Odprava
Razpolovni čas izločanja (t½ v urah) kapecitabina, 5 "-DFCR, 5" -DFUR, 5 -FU in FBAL je bil 0,85 -1,11 -0,66 -0,76 oziroma 3, 23. Kapecitabin in njegovi presnovki se izločajo predvsem v urinu; 95,5% uporabljenega odmerka kapecitabina je bilo izločenega v urinu, izločanje z blatom je minimalno (2,6%). Glavni presnovek, ki se izloča z urinom, je FBLA, ki predstavlja 57% uporabljenega odmerka. Približno 3% uporabljenega odmerka se izloči v urinu kot nespremenjeno zdravilo.
Združena terapija
Študije prve faze, ki so ocenjevale učinke kapecitabina na farmakokinetiko docetaksela ali paklitaksela in obratno, so pokazale, da kapecitabin ne vpliva na farmakokinetiko docetaksela ali paklitaksela (Cmax in AUC) in da docetaksel ali paklitaksel ne vplivata farmakokinetiko 5 "-DFUR.
Farmakokinetika zlasti populacije bolnikov
Po zdravljenju s kapecitabinom v odmerku 1250 mg / m2 dvakrat na dan so pri 505 bolnikih s kolorektalnim rakom opravili populacijsko farmakokinetično analizo. Spol, prisotnost ali odsotnost jetrnih metastaz na začetku, stanje delovanja Karnofskega, skupni bilirubin, serumski albumin, ASAT in ALAT nista statistično pomembno vplivala na farmakokinetiko 5 "-DFUR, 5-FU in FBAL.
Bolniki z okvarjenim delovanjem jeter zaradi jetrnih metastaz: Farmakokinetična študija je pokazala, da se lahko biološka uporabnost kapecitabina in izpostavljenost 5-FU povečata pri bolnikih z rakom z blago do zmerno okvaro jeter zaradi jetrnih metastaz v primerjavi z bolniki brez jetrne okvare. Razpoložljivost farmakokinetičnih podatkov pri bolnikih s hudo okvaro jeter .
Bolniki z okvarjenim delovanjem ledvicNa podlagi rezultatov farmakokinetične študije pri bolnikih z rakom z blago do hudo ledvično okvaro ni dokazov, da očistek kreatinina vpliva na farmakokinetiko matičnega zdravila in 5-FU. Ugotovljeno je bilo, da očistek kreatinina vpliva na "sistemsko izpostavljenost 5" -DFUR (35%povečanje AUC, ko se očistek kreatinina zmanjša za 50%) in FBAL (114%povečanje AUC, ko se očistek kreatinina zmanjša za 50%). 50%). FBAL je presnovek brez antiproliferativne aktivnosti.
Upokojenci: Na podlagi populacijskih farmakokinetičnih analiz, opravljenih pri bolnikih različnih starosti (od 27 do 86 let) in od katerih je bilo 234 (46%) bolnikov starih 65 let ali več, starost ne vpliva na farmakokinetiko 5 "-DFUR in 5 -FU . AUC FBAL se je s starostjo povečeval (20% povečanje starosti vodi do 15% povečanja AUC FBAL). To povečanje je verjetno posledica spremembe v delovanju ledvic.
Etnični dejavniki: Po peroralni uporabi kapecitabina 825 mg / m2 dvakrat na dan 14 dni so imeli japonski bolniki (n = 18) približno 36% nižjo Cmax in 24% nižjo AUC za kapecitabin v primerjavi z belci (n = 22). Japonski bolniki so imeli tudi približno 25% nižjo Cmax in 34% nižjo AUC za FBAL kot belci. Klinični pomen teh razlik ni znan. Pri izpostavljenosti drugim presnovkom (5 "DFCR, 5" DFUR in 5-FU) ni bilo pomembnih razlik.
05.3 Predklinični podatki o varnosti
V študijah toksičnosti pri ponavljajočih se odmerkih je vsakodnevno peroralno dajanje kapecitabina opicam in mišem cynomolgus povzročilo gastrointestinalne, hematopoetske in limfne toksične učinke, značilne za fluoropirimidine. Te strupenosti so bile reverzibilne. Opazili so kožno toksičnost, za katero so značilne degenerativne / regresivne spremembe zaradi kapecitabina. Kapecitabin ni pokazal dokazov o toksičnosti za jetra in centralni živčni sistem. Kardiovaskularna toksičnost (npr. Podaljšanje intervala PR in intervala QT) je bila ugotovljena pri opici cynomolgus po intravenskem dajanju (100 mg / kg), vendar ne po večkratnem odmerjanju (1379 mg / m2 / dan).) Peroralno.
Dvoletna študija rakotvornosti miši ni pokazala dokazov o rakotvornosti zaradi kapecitabina.
V standardnih študijah plodnosti so bile pri samicah miši, ki so jemale kapecitabin, motnje plodnosti; ta učinek pa je bil po obdobju suspenzije zdravila reverzibilen. Poleg tega so bile med 13-tedensko študijo ugotovljene atrofične in degenerativne spremembe v reproduktivnih organih samcev miši; vendar so bili ti učinki po odtegnitvi zdravila reverzibilni (glejte poglavje 4.6).
Študije o embriotoksičnosti in teratogenosti pri miših so pokazale od odmerka odvisno povečanje resorpcije ploda in teratogenosti. Pri opicah so pri visokih odmerkih opazili splav in embrionalno smrtnost, vendar ni bilo nobenih dokazov o teratogenosti.
Kapecitabin ni bil mutagen in vitro za bakterije (Amesov test) ali za celice sesalcev (test mutacije gena kitajskega hrčka V79 / HPRT). Tako kot drugi nukleozidni analogi (t.j. 5-FU) je bil kapecitabin klastogen v človeških limfocitih (in vitro) in je v testu pokazala pozitiven trend (in vivo) mikrojeder v mišičnem kostnem mozgu.
06.0 FARMACEVTSKE INFORMACIJE
06.1 Pomožne snovi
Jedro tabličnega računalnika:
brezvodna laktoza,
natrijeva kroskarmeloza,
hipromeloza,
mikrokristalna celuloza,
magnezijev stearat.
Obloga tablet:
hipromeloza,
titanov dioksid (E171),
rumeni in rdeči železov oksid (E172),
smukec.
06.2 Nezdružljivost
Ni pomembno.
06.3 Obdobje veljavnosti
3 leta.
06.4 Posebna navodila za shranjevanje
Shranjujte pri temperaturi do 30 ° C.
06.5 Vrsta ovojnine in vsebina pakiranja
Narava: PVC / PVDC pretisni omot.
Vsebina: 60 filmsko obloženih tablet (6 pretisnih omotov po 10 tablet).
06.6 Navodila za uporabo in rokovanje
Brez posebnih navodil.
07.0 IMETNIK DOVOLJENJA ZA PROMET
Roche Registration Limited
6 Falcon Way
Park Shire
Vrtno mesto Welwyn
AL7 1TW
UK
08.0 ŠTEVILKA DOVOLJENJA ZA PROMET
EU/1/00/163/001
035219017
09.0 DATUM PRVEGA DOVOLJENJA ALI PODALJŠANJA DOVOLJENJA
Datum prve odobritve: 2. februar 2001
Datum zadnje obnove: 2. februar 2006
10.0 DATUM REVIZIJE BESEDILA
Septembra 2015