Kaj je fenilketonurija
Tam fenilketonurija (P.K.U.) gre za avtosomno recesivno dedno presnovno bolezen, ki prizadene 1 od 10.000 posameznikov in se zdi, da se pojavlja bolj v homozigotnosti kot pri heterozigotih.
Ker spada v skupino hiperfenilalaninemije, fenilketonurija močno ogroža presnovo fenilalanina in zlasti njegovo pretvorbo v tirozin; fenilketonurijo prepoznamo po povišani ravni fenilalanina in nekaterih derivatov v urinu (fenilpiruvat, fenilacetat, fenilaktat in fenilacetilglutamin).
Najresnejši zaplet fenilketonurije je duševna zamuda.
Fenilalanin, tirozin in derivati
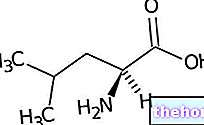
Fenilalanin je esencialna aminokislina in predstavlja večino prehranskih beljakovin; ga lahko encim pretvori fenilalanin hidroksilazo v tirozinu (z dodajanjem hidroksilne skupine -OH). Po drugi strani je tirozin predhodna aminokislina za sintezo:
- L-DOPA (vmesni produkt sinteze dopamina)
- Epinefrin
- Norepinefrin (vsi nevrotransmiterji).

Mehanizem fenilketonurije (P.K.U.)
Kot je bilo pričakovano, je pri fenilketonuriji zaradi ene ali več (vseh 6) kromosomskih mutacij ekspresija (torej presnovna aktivnost) fenilalanin hidroksilaze praktično nič. Te spremembe so lahko različnih vrst (od "missense" sprememb do "spajanja" napak ali celo "delnih izbrisov"), pomembno pa je, da je zaradi te encimske neučinkovitosti koncentracija fenilalanina v krvi (ki je običajno 1 mg / 100 ml) v DOMINANTNI fenilketonuriji zlahka dosežejo količine tudi 50 -krat večje.
Delovanje encima fenilalanin hidroksilaze: Za proizvodnjo tirozina (+ dihidrobiopterina) fenilalanin hidroksilaza potrebuje: fenilalanin, kisik in tetrahidrobiopterin (reducirani pteridin, ki deluje kot hladilni faktor); reakcija je tudi reverzibilna in dihidrobiopterin je mogoče ponovno pretvoriti (zahvaljujoč encimu dihidropterin reduktazo) v tetrahidrobiopterinu.
Zapleti
Fenilketonurija lahko povzroči bolj ali manj hude zaplete glede na resnost patološke manifestacije in pravočasnost diagnoze; kot dedna patologija se fenilketonurija razlikuje po:
- Prevladujoča, zato je značilna POPOLNA neaktivnost encima fenilalanin hidroksilaze
- Recesivno, pri katerem je aktivnih le 30% celotne encimske dediščine.
Zaplete fenilketonurije je mogoče pripisati in sorazmerno sorazmerno s presnovno kopičenjem fenilalanina, njegovih derivatov in zmanjšano sintezo tirozina. Pri patologiji presežek fenilalanina relativno učinkovito filtrira ledvica, ki ga le delno absorbira in ga odstrani z urinom ; vendar obstojnost ravni hiperfenilalaninemije določa presnovno reakcijo molekularne PREVERJANJA v fenilpiruvična kislina in / ali druge derivate, ki jih je lažje odvajati (fenilpiruvat, fenilacetat, fenilaktat).
Fenilketonurijo otežuje toksičnost fenilalanina, fenilpiruvične kisline in njenih derivatov proti centralnemu živčnemu sistemu (CNS). Njihova prekomerna prisotnost v razvoju možganov neizprosno določa obliko duševne zaostalosti.
Opomba: Plazemske koncentracije drugih aminokislin so rahlo znižane, verjetno zaradi povratnih informacij o črevesni absorpciji ali reabsorpciji ledvičnih tubulov.
Poškodbe možganov kot resen zaplet fenilketonurije povzročijo odštevanje drugih esencialnih aminokislin pri proteosintezi, zlasti pri tvorbi poliribosomov, mielina, noradrenalina in serotonina. Fenilketonurija - ni vidna takoj po rojstvu, ampak po nekaj letih - če se ne zdravi, zahteva hospitalizacijo otroka in je popolnoma nepopravljiva.
Napredna fenilketonurija je lahko jasno vidna tudi s prostim očesom; visoke koncentracije fenilalanina, ki zavira encim tirozinaza, znatno zmanjšajo sintezo melanina z zmanjšanjem pigmentacije kože in las; poleg tega kopičenje fenilacetata v laseh in koži daje fenilketonurici močan in neprijeten "vonj po miših".