Kaj je Marfanov sindrom?
Marfanov sindrom opisuje kompleksno dedno motnjo vezivnega tkiva, ki prizadene predvsem oči, srčno -žilni sistem in mišično -skeletni sistem. Ker pa je vsak organ sestavljen iz vezivnega tkiva, lahko Marfanov sindrom v idealnem primeru uniči in močno moti rast in delovanje katerega koli anatomskega mesta.
Sindrom se prenaša kot avtosomno dominantna lastnost: zato se soočamo z resno genetsko boleznijo, ki ima izjemno spremenljivo "fenotipsko izražanje (okvare se lahko od družine do družine ali od bolnika do bolnika zelo razlikujejo)".
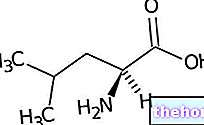
Marfanov sindrom sproži sprememba gena FBN1 (na kromosomu 15), ki kodira fibrilin-1, zelo pomemben vezni glikoprotein, ki predstavlja strukturno podporo za mikrofibrile.
Mikrofibrili: sestavljeni iz fibrilina, so mikrofibrili prisotni v zunajceličnem matriksu, v katerem tvorijo preplet za odlaganje elastina v elastičnih vlaknih. Čeprav so v telesu povsod prisotne, mikrofibrile obilujejo predvsem v aorti, ligamentih in zonah ciliarna telesa (na očesni ravni).
Ker gre za avtosomno dominantno bolezen, le Marfanov sindrom prizadene samo otroke, ki so podedovali spremenjen gen FBN-1 od obeh staršev. Kljub temu je v enem od štirih primerov bolezen posledica spontanih mutacij pri bolnikih, ki nimajo družinske anamneze.
Ime bolezni izvira iz francoskega pediatra, ki jo je prvič opisal leta 1896 (A. Marfan), nato pa je bilo treba počakati do leta 1991, da bi ugotovili spremenjeni gen, ki je bil vključen v simptomatološko manifestacijo: odkritelj je bil F. Ramirez.
Poglej si posnetek
- Oglejte si video na youtube
Vzroki
Omenili smo, da je Marfanov sindrom takojšen izraz mutacije gena, ki kodira fibrilin-1.
FIBRILLIN 1 je glikoproteinska komponenta elastina, ki je bistvena za zagotavljanje in vzdrževanje elastičnosti in trdnosti tkiva.V fizioloških pogojih se fibrilin 1 veže na drugo beljakovino, znano kot TGF-beta (ali transformacijski rastni faktor beta). Zdi se, da je TGF-beta vključen v škodljive procese, ki vplivajo na gladke mišice krvnih žil in zunajcelični matriks. Na podlagi teh predpostavk so nekateri avtorji prepričani, da je Marfanov sindrom poleg mutacije gena FBN-1 tudi presežek TGF-beta, zlasti v aorti, srčnih zaklopkah in pljučih.

Incidenca
Ocenjuje se, da Marfanov sindrom prizadene 1 od 3.000 do 5.000 rojstev in se pojavlja brez razlikovanja med moškimi in ženskami. Statistični podatki kažejo, da ima 75% bolnikov pozitivno družinsko anamnezo; v preostalih 25% vzrok ležijo v občasnih mutacijah, za katere se zdi, da so na nek način povezane z očetovo starostjo v trenutku spočetja.
Otroci z izjemno hudimi oblikami Marfanovega sindroma imajo "pričakovano življenjsko dobo manj kot eno leto".
Pred razvojem operativnih strategij na odprtem srcu je imela večina bolnikov z Marfanovim sindromom povprečno pričakovano življenjsko dobo 32 let; zahvaljujoč nenehnemu izboljševanju medicinske in farmakološke terapije trenutno bolniki z Marfanovim sindromom živijo povprečno do 60 let.
Znaki in simptomi
Za dodatne informacije: simptomi Marfanovega sindroma
Marfanov sindrom se lahko pojavi popolnoma asimptomatsko. Prizadeti bolniki imajo pretirano vitko strukturo, saj so nesorazmerno visoki in tanki. Spodnje in zgornje okončine so veliko daljše od trupa (dolikostenomegalija). Govori se tudi o arahnodaktilija kar najbolje izraziti koncept pretirane dolžine prstov, značilen za tiste, ki jih prizadene Marfanov sindrom: roke se zato primerjajo z nogami pajka.
Kar zadeva višino, imajo ti bolniki rast s povprečjem nad 97. percentilom.
Med drugimi značilnostmi, ki so pogosto prisotne pri bolnikih z Marfanovim sindromom, se spomnimo tudi:
- Odpiranje rok večje od višine
- Ohlapni sklepi → pretirana gibljivost sklepov
- Deformacija prsne stene
- Premik leče
- Zgornji del telesa je manj razvit kot spodnji del
- Spontani pnevmotoraks (11%)
- Skolioza
- Kožne strije na ravni stegna, hrbta, deltoidne, prsne
Med najbolj problematičnimi znaki, povezanimi z Marfanovim sindromom, se spomnimo prolapsa srčne zaklopke in insuficience mitralne zaklopke: podobno stanje lahko zlahka spodbudi razširitev aortnega obroča in disekcijo aorte.
Tabela prikazuje znake, ki jih lahko najdemo pri bolnikih z Marfanovim sindromom. Tam opisani liki niso vedno prisotni, a dober del jih je mogoče najti.
Možni simptomi
Koža
Strije v prsnem, ledvenem in križnem predelu
Oči
Sprememba vida, astigmatizem, odmik mrežnice, glavkom zaprtega kota, luksacija leče, kratkovidnost
Kostna struktura
Artralgija, kifoskolioza, dolihostenomelija (prekomerni razvoj v dolžini udov glede na trup), hipermobilnost, visoko nebo, deformirane prsi, ploska stopala, tesna in tanka zapestja, nenormalen ponovni vstop / izboklina prsnice, skolioza, ukrivljena ramena, spondilolisteza
Prsti
arahnodaktilija
Pljuča
Spontani pnevmotoraks, dispneja, idiopatska obstruktivna pljučna bolezen
Obrazne spremembe
Ogivalno nebo (malformacija neba), mandibularna retrognatija (razvojna napaka čeljusti), podolgovat obraz
Srce
Angina pektoris, anevrizma trebušne aorte, srčna aritmija, razširitev / ruptura / disekcija prsne aorte, aortna insuficienca, prolaps mitralne zaklopke
Jezik
Težave z govorom
Diagnoza
Glede na več kot 200 možnih mutacij je uporaba genetskih označevalcev v diagnostične namene skoraj nemogoča.
Ocena Marfanovega sindroma ni vedno tako neposredna, saj fenotipski izraz mutacije ni vedno očiten in ga je enostavno prepoznati. Diagnostična zamuda lahko resno ogrozi bolnikovo preživetje: pomislite na primer na to, da ne prepoznate srčno -žilne težave.
Diagnostična merila za Marfanov sindrom so bila mednarodno oblikovana leta 1996: diagnoza je sestavljena iz "preiskave družinske anamneze, povezane s kombinacijo glavni in manjši kazalniki sindroma.
Nekateri številni uporabljeni diagnostični testi so:
- ehokardiogram
- magnetna angioresonanca in CT (za preiskavo aorte)
- angiografija z magnetno resonanco (MRA) s kontrastno tekočino (za poudarjanje notranjih struktur aorte)
- pregled z režami (za analizo možne dislokacije leče)
- merjenje očesnega tlaka (za poudarjanje možne prisotnosti glavkoma)
- genetski testi (priporočljivo pred spočetjem otroka, da se ugotovi, ali je sindrom ali ne)
Terapije
Ker gre za genetsko bolezen, ni nobenega zdravila ali zdravljenja, ki bi bolezen lahko obrnilo.
Uporaba zdravil pa je ključnega pomena za lajšanje simptomov in izogibanje kakršnim koli zapletom, zlasti srčnim. V ta namen so še posebej primerna zdravila za zniževanje krvnega tlaka, na primer sartani (predvsem), zaviralci ACE in zaviralci adrenergičnih receptorjev beta.
V okviru Marfanovega sindroma lahko bolniki, ki trpijo tudi za skoliozo, sledijo posebnemu zdravljenju, pa tudi pri bolnikih z glavkomom.
Operacija je možna za odpravo nenormalne dilatacije aorte, ki pogosto združuje večino bolnikov z Marfanovim sindromom.
Nadaljuj: Marfanov sindrom - zdravila in zdravljenje "