Aktivne sestavine: Pegfilgrastim
Neulasta 6 mg raztopina za injiciranje v napolnjeni injekcijski brizgi
Indikacije Zakaj se zdravilo Neulasta uporablja? Za kaj je to?
Neulasta vsebuje zdravilno učinkovino pegfilgrastim. Pegfilgrastim je protein, proizveden z biotehnološko tehniko v bakterijski celici, imenovani Escherichia coli. Spada v skupino beljakovin, imenovanih citokini, in je zelo podoben naravnemu proteinu (faktorju, ki stimulira kolonije granulocitov), ki ga proizvaja naše telo.
Zdravilo Neulasta se uporablja za zmanjšanje trajanja nevtropenije (nizko število belih krvnih celic) in pojav vročinske nevtropenije (nizko število belih krvnih celic z zvišano telesno temperaturo), ki jo lahko povzroči citotoksična kemoterapija (zdravila, ki uničujejo hitro rastoče celice). Bele krvne celice so pomembne, ker pomagajo telesu pri boju proti okužbam. Te celice so zelo občutljive na učinke kemoterapije, kar lahko povzroči zmanjšanje števila teh celic v telesu. Če število belih krvnih celic pade na nizko raven, morda ne bo dovolj za boj proti bakterijam in obstaja večja nevarnost okužbe.
Zdravnik vam je predpisal zdravilo Neulasta, da stimulira vaš kostni mozeg (del kosti, ki tvori krvne celice), da ustvari več belih krvnih celic, ki pomagajo telesu pri boju proti okužbam.
Kontraindikacije Ko se zdravila Neulasta ne sme uporabljati
Ne uporabljajte zdravila Neulasta, če ste alergični na pegfilgrastim, filgrastim, beljakovine, pridobljene iz Escherichia coli, ali katero koli drugo sestavino tega zdravila.
Previdnostni ukrepi pri uporabi Kaj morate vedeti, preden boste vzeli zdravilo Neulasta
Pred uporabo zdravila Neulasta se posvetujte s svojim zdravnikom, farmacevtom ali medicinsko sestro:
- če imate alergijsko reakcijo, vključno s šibkostjo, padcem krvnega tlaka, težavami z dihanjem, otekanjem obraza (anafilaksa), pordelostjo in pordelostjo, kožnim izpuščajem in srbečimi deli kože
- če ste alergični na lateks. Pokrovček igle napolnjene injekcijske brizge vsebuje derivat lateksa, ki lahko povzroči hude alergijske reakcije
- če imate kašelj, zvišano telesno temperaturo in težave z dihanjem. To je lahko znak sindroma akutne respiratorne stiske (ARDS).
- če imate enega ali več naslednjih neželenih učinkov:
- otekanje ali otekanje, ki je lahko povezano z manj tekočine, oteženo dihanje, napihnjenost v trebuhu in občutek sitosti ter splošen občutek utrujenosti To so lahko simptomi stanja, imenovanega "sindrom kapilarnega puščanja" telo. Glej odstavek 4.
- če imate bolečine v levem zgornjem delu trebuha ali bolečine v okončini rame. To so lahko znaki težave z vranico (splenomegalija)
- če ste pred kratkim imeli "hudo pljučno okužbo (pljučnica), tekočino v pljučih (pljučni edem), vnetje pljuč (intersticijska pljučna bolezen)" ali "nenormalnost, ugotovljeno na rentgenskih žarkih (pljučna infiltracija)
- če veste, da imate nenormalno število krvnih celic (npr. povečano število belih krvnih celic ali anemijo) ali znižanje ravni trombocitov, kar zmanjša sposobnost strjevanja telesa (trombocitopenija). Zdravnik vas bo morda želel pozorno spremljati
- če imate srpastocelično anemijo. Zdravnik vas bo morda želel pozorno spremljati
- če imate nenadoma znake alergije, kot so izpuščaj, koprivnica ali srbeča koža, otekanje obraza, ustnic, jezika ali drugih delov telesa, težko dihanje, piskanje ali težko dihanje, so to lahko znaki resne alergijske reakcije.
Zdravnik vam bo redno pregledoval kri in urin, saj lahko zdravilo Neulasta poškoduje drobne filtre v ledvicah (glomerulonefritis).
O tveganjih za nastanek krvnega raka se morate pogovoriti s svojim zdravnikom. Če imate ali imate morda krvni rak, zdravila Neulasta ne smete uporabljati, razen če vam tako naroči zdravnik.
Izguba odziva na pegfilgrastim
Če ste zmanjšali odziv ali ne vzdržujete odziva na zdravljenje s pegfilgrastimom, bo zdravnik preiskal razloge, vključno z možnostjo, da ste razvili protitelesa, ki nevtralizirajo aktivnost pegfilgrastima.
Interakcije Katera zdravila ali živila lahko spremenijo učinek zdravila Neulasta
Povejte svojemu zdravniku ali farmacevtu, če jemljete, ste pred kratkim jemali ali pa boste morda začeli jemati katero koli drugo zdravilo.
Opozorila Pomembno je vedeti, da:
Nosečnost in dojenje
Preden vzamete katerokoli zdravilo, se posvetujte z zdravnikom ali farmacevtom. Zdravila Neulasta niso testirali pri nosečnicah. Pomembno je, da zdravniku poveste, če:
- ste noseči;
- sum nosečnosti; ali
- načrtuje nosečnost.
Če med zdravljenjem z zdravilom Neulasta zanosite, obvestite svojega zdravnika. Morda boste vabljeni, da se vpišete v Amgenov program spremljanja nosečnosti. Kontaktni podatki lokalnega predstavnika so navedeni v poglavju 6 te brošure.
Če vam zdravnik ne pove drugače, morate z uporabo zdravila Neulasta prenehati z dojenjem.
Če med jemanjem zdravila Neulasta dojite, vas lahko spodbudimo, da se vključite v program Amgenovega nadzora dojenja.
Vpliv na sposobnost upravljanja vozil in strojev
Neulasta nima vpliva ali ima zanemarljiv vpliv na sposobnost vožnje in upravljanja s stroji.
Neulasta vsebuje sorbitol (E420) in natrijev acetat
Neulasta vsebuje sorbitol (vrsto sladkorja). Če vam je zdravnik povedal, da ne prenašate nekaterih sladkorjev, se pred jemanjem tega zdravila posvetujte z zdravnikom.
To zdravilo vsebuje manj kot 1 mmol (23 mg) natrija na odmerek 6 mg, v bistvu je brez natrija.
Odmerjanje, način in čas dajanja Kako uporabljati zdravilo Neulasta: Odmerjanje
Neulasta je indicirana pri odraslih, starih 18 let ali več.
Pri jemanju zdravila Neulasta natančno upoštevajte zdravnikova navodila. Če ste v dvomih, se posvetujte z zdravnikom ali farmacevtom. Običajni odmerek je podkožna injekcija (injekcija pod kožo) 6 mg z napolnjeno injekcijsko brizgo, ki jo je treba dati vsaj 24 ur po zadnjem odmerku kemoterapije na koncu vsakega ciklusa kemoterapije.
Neulaste močno ne pretresite, saj lahko to ogrozi njeno delovanje.
Kako si injicirati zdravilo Neulasta
Zdravnik bo morda menil, da je najbolje, da si zdravilo Neulasta injicirate sami. Zdravnik ali medicinska sestra vam bosta pokazala, kako si injicirati zdravilo Neulasta. Ne poskušajte si injicirati sami, če vam ni bilo naročeno, kako injicirati.
Navodila, kako si zdravilo Neulasta injicirate sami, preberite v razdelku na koncu tega navodila.
Če ste pozabili na injiciranje zdravila Neulasta
Če ste pozabili na odmerek zdravila Neulasta, se obrnite na svojega zdravnika, da ugotovi, kdaj boste dobili naslednjo injekcijo.
Če imate dodatna vprašanja o uporabi tega zdravila, se posvetujte z zdravnikom, farmacevtom ali medicinsko sestro.
Preveliko odmerjanje Kaj storiti, če ste vzeli preveč zdravila Neulasta
Če ste uporabili večji odmerek zdravila Neulasta, kot bi smeli, se obrnite na svojega zdravnika, farmacevta ali medicinsko sestro.
Neželeni učinki Kakšni so neželeni učinki zdravila Neulasta
Kot vsa zdravila ima lahko tudi to zdravilo neželene učinke, ki pa se ne pojavijo pri vseh bolnikih.
Takoj obvestite svojega zdravnika, če opazite katerega od naslednjih neželenih učinkov ali kombinacijo teh neželenih učinkov:
- otekanje ali otekanje, ki je lahko povezano z redkejšim prehajanjem vode, oteženo dihanje, napihnjenost in občutek sitosti ter splošen občutek utrujenosti. Ti simptomi se običajno hitro razvijejo.
To so lahko simptomi občasnega stanja (pojavijo se lahko pri največ 1 od 100 ljudi), imenovanega "sindrom puščanja kapilar", ki povzroči uhajanje krvi iz majhnih krvnih žil v telo in potrebuje nujno zdravniško pomoč.
Zelo pogosti neželeni učinki (pojavijo se lahko pri več kot 1 od 10 bolnikov):
- bolečine v kosteh. Zdravnik vam bo povedal, kaj morate vzeti za lajšanje bolečin v kosteh.
- slabost in glavobol.
Pogosti neželeni učinki (pojavijo se lahko pri največ 1 od 10 bolnikov):
- bolečine na mestu injiciranja.
- splošne bolečine v sklepih in mišicah.
- v krvi se lahko pojavijo nekatere spremembe, ki pa bodo zaznane med rutinskimi preiskavami krvi. Ravni belih krvnih celic se lahko za kratek čas zvišajo. Raven trombocitov lahko pade in povzroči modrice.
Občasni neželeni učinki (pojavijo se lahko pri največ 1 od 100 bolnikov):
- reakcije alergijskega tipa, vključno z rdečico in zardevanjem, kožnim izpuščajem (pordelost kože) in srbečim otekanjem kože.
- hude alergijske reakcije, vključno z anafilaksijo (šibkost, padec krvnega tlaka, težave z dihanjem, otekanje obraza).
- povečanje volumna vranice.
- ruptura vranice. Nekateri primeri razpokane vranice so bili usodni. Pomembno je, da se takoj obrnete na svojega zdravnika, če čutite bolečino v zgornji levi strani trebuha ali levi rami, saj lahko to kaže na težave z vranico.
- težave z dihanjem. Če imate kašelj, zvišano telesno temperaturo in težave z dihanjem, se posvetujte z zdravnikom.
- so bili primeri Sweetovega sindroma (vijolične, dvignjene in boleče lezije na okončinah in včasih na obrazu in vratu, povezane z zvišano telesno temperaturo), lahko pa so prispevali tudi drugi dejavniki
- kožni vaskulitis (vnetje kožnih krvnih žil).
- poškodbe drobnih filtrov v ledvicah (glomerulonefritis).
- pordelost na mestu injiciranja.
Poročanje o stranskih učinkih
Če opazite kateri koli neželeni učinek, se posvetujte z zdravnikom, farmacevtom ali medicinsko sestro, kar vključuje morebitne neželene učinke, ki niso navedeni v tem navodilu.
Potek in zadržanje
Zdravilo shranjujte nedosegljivo otrokom!
Tega zdravila ne smete uporabljati po datumu izteka roka uporabnosti, ki je naveden na škatli in brizgi poleg oznake EXP. Rok uporabnosti se nanaša na zadnji dan navedenega meseca.
Shranjujte v hladilniku (2 ° C - 8 ° C).
Neulasto lahko vzamete iz hladilnika in jo hranite pri sobni temperaturi (največ 30 ° C) največ 3 dni. Ko brizgo odstranite iz hladilnika in doseže sobno temperaturo (ne višjo od 30 ° C), jo morate uporabiti v 3 dneh ali jo zavreči.
Ne zamrzujte. Neulasta se lahko uporablja, če je bila po nesreči enkrat zamrznjena manj kot 24 ur.
Vsebnik shranjujte v zunanji ovojnini, da zdravilo zaščitite pred svetlobo.
Tega zdravila ne uporabljajte, če opazite, da je motno ali vidite delce.
Ne mečite nobenih zdravil v odpadne vode ali med gospodinjske odpadke. Vprašajte svojega farmacevta, kako zavreči zdravila, ki jih ne uporabljate več. Tako boste zaščitili okolje.
Rok "> Druge informacije
Kaj vsebuje zdravilo Neulasta
- Zdravilna učinkovina je pegfilgrastim. Vsaka napolnjena injekcijska brizga vsebuje 6 mg pegfilgrastima v 0,6 ml raztopine.
- Pomožne snovi so natrijev acetat, sorbitol (E420), polisorbat 20 in voda za injekcije. Glej odstavek 2.
Izgled zdravila Neulasta in vsebina pakiranja
Neulasta je bistra, brezbarvna raztopina za injiciranje v napolnjeni injekcijski brizgi (6 mg / 0,6 ml).
Vsako pakiranje vsebuje 1 napolnjeno injekcijsko brizgo iz stekla tipa I. z iglo iz nerjavečega jekla in pokrovčkom igle. Brizge so pakirane v pretisnem omotu ali brez pretisnega omota.
Expiry "> Navodila za injiciranje napolnjene injekcijske brizge Neulasta
Ta razdelek vsebuje informacije o tem, kako si sami injicirate zdravilo Neulasta.
Pomembno je, da si ne poskušate injicirati sami, če vam zdravnik, medicinska sestra ali farmacevt niso povedali, kako to storiti. Če imate kakršna koli vprašanja o injiciranju, se za pomoč obrnite na svojega zdravnika, medicinsko sestro ali farmacevta.
Kako uporabljati zdravilo Neulasta v napolnjeni injekcijski brizgi vi ali oseba, ki vam daje injekcijo
Injicirati se boste morali pod kožo, kar se imenuje podkožno.
Kaj je potrebno
Za "podkožno injekcijo" boste potrebovali:
- ena napolnjena injekcijska brizga zdravila Neulasta; In
- alkoholne robčke ali podobna razkužila.
Kaj naj storim, preden si dam podkožno injekcijo zdravila Neulasta?
- Zdravilo vzemite iz hladilnika.
- Napolnjene injekcijske brizge ne tresite.
- Pokrovčka igle ne odstranite z brizge, dokler niste pripravljeni na injiciranje.
- Preverite rok uporabnosti na nalepki napolnjene injekcijske brizge (EXP). Ne uporabljajte je po zadnjem dnevu v prikazanem mesecu.
- Preverite videz zdravila Neulasta. Mora biti bistra, brezbarvna tekočina. Če vidite delce, ga ne smete uporabiti.
- Za bolj udobno injiciranje pustite napolnjeno injekcijsko brizgo pol ure iz hladilnika, da doseže sobno temperaturo, ali pa jo nekaj minut nežno držite v roki. Neulaste ne ogrevajte na noben drug način (npr. Ne segrevajte v mikrovalovni pečici ali v vroči vodi).
- Temeljito si umijte roke.
- Poiščite udobno, dobro osvetljeno in čisto površino in imejte pri roki vse, kar potrebujete.
Kako pripravim injekcijo zdravila Neulasta?
Preden si injicirate zdravilo Neulasta, morate narediti naslednje:
- Brizgo vzemite v roko in nežno odstranite pokrovček z igle, ne da bi ga upognili. Povlecite vodoravno. Ne dotikajte se igle in ne pritiskajte bata.
- V napolnjeni injekcijski brizgi lahko opazite majhen zračni mehurček. Pred injiciranjem ne smete odstraniti zračnega mehurčka. Injiciranje raztopine z zračnim mehurčkom je neškodljivo.
- Zdaj lahko uporabite napolnjeno injekcijsko brizgo.
Kje naj si dam injekcijo?
Najprimernejša mesta za injiciranje so:
- zgornji del stegen; In
- trebuh, razen območja okoli popka.
Če vam bo kdo drug dal injekcijo, lahko uporabite tudi zadnji del rok.
Kako si dam injekcijo?
- Kožo očistite z alkoholno krpico.
- Dvignite kožo med palcem in kazalcem (ne da bi ga stisnili). Iglo potisnite v kožo.
- Bat počasi in enakomerno pritiskajte navzdol. Bat potisnite do konca, dokler ne vbrizgate vse tekočine.
- Po injiciranju tekočine izvlecite iglo in spustite kožo.
- Če na mestu injiciranja opazite majhno kapljico krvi, jo nežno obrišite z vato ali gazo. Ne drgnite mesta injiciranja. Po potrebi lahko mesto injiciranja prekrijete z obližem.
- Ostanka zdravila Neulasta v brizgi ne uporabljajte ponovno.
Spomniti se
Vsako brizgo uporabite samo za eno injekcijo. Če imate težave, se za pomoč in nasvet obrnite na svojega zdravnika ali medicinsko sestro.
Odstranitev rabljenih brizg
- Pokrovčka ne nameščajte nazaj na uporabljene igle.
- Uporabljene brizge shranjujte nedosegljivo otrokom!
- Uporabljene brizge je treba odstraniti v skladu z lokalnimi predpisi. Vprašajte svojega farmacevta, kako zavreči zdravila, ki jih ne uporabljate več. To bo pomagalo zaščititi okolje.
Navodilo za uporabo vira: AIFA (Italijanska agencija za zdravila). Vsebina, objavljena januarja 2016. Prisotne informacije morda niso posodobljene.
Za dostop do najnovejše različice je priporočljivo dostopati do spletnega mesta AIFA (Italijanska agencija za zdravila). Zavrnitev odgovornosti in koristne informacije.
01.0 IME ZDRAVILA -
NEULASTA 6 MG raztopina za injiciranje
02.0 KAKOVOSTNA IN KOLIČINSKA SESTAVA -
Vsaka napolnjena injekcijska brizga vsebuje 6 mg pegfilgrastima * v 0,6 ml raztopine za injiciranje. Koncentracija je 10 mg / ml, upoštevajoč le delež beljakovin **.
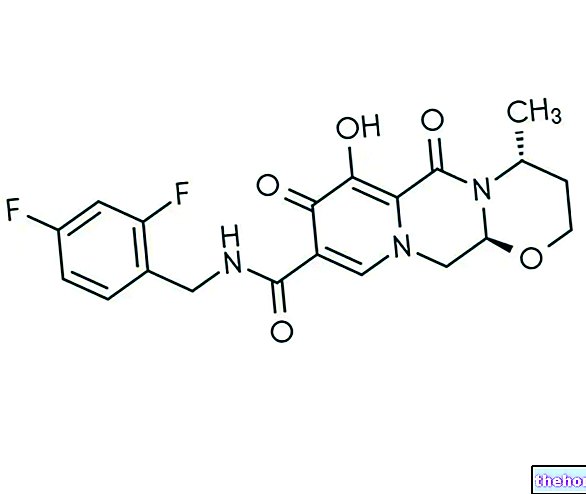
* Pegfilgrastim se proizvaja v celicah Escherichia coli s tehnologijo rekombinantne DNA in naknadno konjugacijo s polietilen glikolom (PEG).
** Koncentracija je 20 mg / ml, če je vključen del molekule PEG.
Učinkovitosti tega izdelka ne smemo primerjati z drugimi pegiliranimi ali nepegiliranimi beljakovinami iz istega terapevtskega razreda.
Za dodatne informacije glejte poglavje 5.1.
Pomožne snovi z znanim učinkom:
Vsaka napolnjena injekcijska brizga vsebuje 30 mg sorbitola (E420)
Vsaka napolnjena injekcijska brizga vsebuje manj kot 1 mmol (23 mg) natrija (glejte poglavje 4.4).
Za celoten seznam pomožnih snovi glejte poglavje 6.1.
03.0 FARMACEVTSKA OBLIKA -
Raztopina za injiciranje.
Bistra in brezbarvna raztopina za injiciranje.
04.0 KLINIČNE INFORMACIJE -
04.1 Terapevtske indikacije -
Zmanjšanje trajanja nevtropenije in pojavnosti febrilne nevtropenije pri odraslih bolnikih, zdravljenih s citotoksično kemoterapijo za raka (z izjemo kronične mieloične levkemije in mielodisplastičnih sindromov).
04.2 Odmerjanje in način uporabe -
Zdravljenje z zdravilom Neulasta morajo začeti in nadzorovati zdravniki z onkološko in / ali hematološko izkušnjo.
Odmerjanje
Za vsak cikel kemoterapije je priporočljiv odmerek 6 mg (ena napolnjena injekcijska brizga) zdravila Neulasta, ki se daje vsaj 24 ur po citotoksični kemoterapiji.
Način dajanja
Zdravilo Neulasta se injicira subkutano. Injekcijo je treba dati v stegno, trebuh ali nadlaket. Za navodila o ravnanju z zdravilom pred uporabo glejte poglavje 6.6.
Pediatrična populacija
Varnost in učinkovitost zdravila Neulasta pri otrocih še nista bili ugotovljeni, trenutno razpoložljivi podatki so opisani v poglavjih 4.8, 5.1 in 5.2, vendar ni mogoče dati priporočil o odmerjanju.
Bolniki z okvaro ledvic
Pri bolnikih z okvaro ledvic, vključno s tistimi s končno odpovedjo ledvic, prilagoditev odmerka ni priporočljiva.
04.3 Kontraindikacije -
Preobčutljivost za zdravilno učinkovino ali katero koli pomožno snov.
04.4 Posebna opozorila in ustrezni previdnostni ukrepi pri uporabi -
Omejeni klinični podatki kažejo na primerljiv učinek pegfilgrastima v primerjavi s filgrastimom na čas do remisije hude nevtropenije pri bolnikih z akutno mieloično levkemijo. de novo (glejte poglavje 5.1). Vendar dolgoročni učinki zdravila Neulasta pri akutni mieloični levkemiji niso bili ugotovljeni; zato je treba pri tej populaciji bolnikov zdravilo uporabljati previdno.
Faktor, ki stimulira kolonije granulocitov, lahko spodbuja rast mieloidnih celic in vitro in podobne učinke lahko opazimo in vitro v nekaterih ne-mieloidnih celicah.
Varnost in učinkovitost zdravila Neulasta nista bili raziskani pri bolnikih z mielodisplastičnim sindromom, kronično mieloično levkemijo in pri bolnikih s sekundarno akutno mieloično levkemijo (AML), zato ga pri teh bolnikih ne smemo uporabljati. diagnoza blastne transformacije kronične mieloične levkemije iz akutne mieloične levkemije.
Učinkovitost in varnost uporabe zdravila Neulasta pri bolnikih z AML de novo starosti
Varnost in učinkovitost zdravila Neulasta pri bolnikih, ki so prejemali kemoterapijo z visokimi odmerki, nista bili raziskani.
Neželeni učinki na pljučih
Po dajanju G-CSF so poročali o občasnih pljučnih neželenih učinkih (≥ 1 / 1.000, intersticijski pljučnici.) Bolniki z nedavno anamnezo pljučnih infiltratov ali pljučnice so lahko bolj ogroženi (glejte poglavje 4.8).
Začetek pljučnih simptomov, kot so kašelj, zvišana telesna temperatura in dispneja hkrati z radiološko sliko pljučnih infiltratov in poslabšanjem delovanja pljuč, povezano s povišanim številom belih krvnih celic, je lahko začetni znak sindroma akutne respiratorne stiske (Akutni respiratorni distresni sindrom, ARDS). V takih okoliščinah je po presoji zdravnika treba zdravljenje z zdravilom Neulasta prekiniti in uvesti ustrezno zdravljenje (glejte poglavje 4.8).
Glomerulonefritis
Pri bolnikih, ki so prejemali filgrastim in pegfilgrastim, so poročali o glomerulonefritisu. Na splošno so dogodki glomerolunefritisa izginili po zmanjšanju odmerka ali prekinitvi zdravljenja s filgrastimom in pegfilgrastimom. Priporočljivo je spremljanje urina.
Sindrom puščanja kapilar
Po dajanju faktorjev, ki stimulirajo kolonije granulocitov, so poročali o sindromu kapilarnega puščanja, za katerega so značilne hipotenzija, hipoalbuminemija, edemi in hemokoncentracija. Bolnike, pri katerih se pojavijo simptomi sindroma puščanja kapilar, je treba skrbno spremljati in prejeti standardno simptomatsko zdravljenje, ki lahko vključuje potrebo po intenzivni negi (glejte poglavje 4.8).
Splenomegalija in ruptura vranice
Po dajanju pegfilgrastima so poročali o občasnih, vendar na splošno asimptomatskih primerih splenomegalije in občasnih primerih rupture vranice, vključno z nekaterimi smrtnimi primeri (glejte poglavje 4.8). Zato je treba skrbno spremljati volumen vranice (npr. S kliničnim pregledom, ultrazvokom). Pri bolnikih z bolečino v trebuhu ali rami v levem zgornjem kvadrantu je treba razmisliti o diagnozi vranice.
Trombocitopenija in anemija
Zdravljenje samo z zdravilom Neulasta ne izključuje trombocitopenije in anemije, ki sta posledica vzdrževanja polnih odmerkov mielosupresivne kemoterapije. Priporoča se redno spremljanje števila trombocitov in hematokrita. Posebno pozornost je treba nameniti pri dajanju posameznih ali kombiniranih kemoterapevtikov, ki povzročajo hudo trombocitopenijo.
Anemija srpastih celic
Krize srpastih celic so bile povezane z uporabo pegfilgrastima pri bolnikih s srpastoceličnimi lastnostmi ali boleznijo srpastih celic (glejte poglavje 4.8) .Zato mora biti zdravnik previden pri predpisovanju zdravila Neulasta bolnikom s srpastoceličnimi lastnostmi ali srpastoceličnimi boleznimi, hranite ustrezne klinične in laboratorijske parametre, pri tem pa bodite pozorni na možno povezavo med tem zdravilom in povečano vranico ter vazookluzivno krizo.
Levkocitoza
Vrednosti belih krvnih celic (Bele krvne celice(WBC), enakih ali večjih od 100 x 109 / l, so opazili pri manj kot 1% bolnikov, zdravljenih z zdravilom Neulasta. , običajno opazimo 24 - 48 ur po dajanju in je skladen s farmakodinamičnimi učinki tega zdravila.V skladu s kliničnimi učinki in možnostjo levkocitoze je treba med zdravljenjem v rednih časovnih presledkih izvajati število belih krvnih celic (WBC). če število belih krvnih celic preseže 50 x 109 / L po pričakovani vrednosti, je treba dajanje tega zdravila takoj prekiniti.
Preobčutljivost
Pri bolnikih, zdravljenih z zdravilom Neulasta, so poročali o preobčutljivostnih reakcijah, vključno z anafilaktičnimi reakcijami, ki so se pojavile na začetku ali po zdravljenju. Zdravljenje z zdravilom Neulasta pri bolnikih s klinično pomembno preobčutljivostjo trajno prekinite. Ne dajajte zdravila Neulasta bolnikom z anamnezo preobčutljivosti na pegfilgrastim ali filgrastim Če pride do hude alergijske reakcije, je treba dati ustrezno terapijo, nato pa bolnika skrbno spremljati več dni.
Imunogenost
Tako kot pri vseh terapevtskih beljakovinah obstaja potencialno tveganje imunogenosti. Verjetnost nastanka protiteles proti pegfilgrastimu je na splošno majhna. Pri vseh bioloških zdravilih se pričakuje razvoj vezavnih protiteles, ki pa do danes niso bila povezana z aktivnostjo. Nevtralizacija.
Varnost in učinkovitost zdravila Neulasta pri mobilizaciji hematopoetskih matičnih celic pri zdravih bolnikih ali darovalcih nista bili ustrezno ocenjeni.
Pokrovček igle napolnjene injekcijske brizge vsebuje suh naravni kavčuk (derivat lateksa), ki lahko povzroči alergijske reakcije.
Povečana hematopoetska aktivnost kostnega mozga kot odziv na terapijo z rastnim faktorjem je bila povezana s prehodno pozitivnimi radiološkimi ugotovitvami kosti, kar je treba upoštevati pri razlagi radioloških podatkov.
Neulasta vsebuje sorbitol. Bolniki z redko dedno intoleranco za fruktozo ne smejo jemati tega zdravila.
Neulasta vsebuje manj kot 1 mmol (23 mg) natrija v odmerku 6 mg, kar pomeni, da v bistvu "ne vsebuje natrija".
Da bi izboljšali sledljivost faktorjev, ki stimulirajo kolonije granulocitov (G-CSF), je treba trgovsko ime zdravila, ki se daje, jasno zabeležiti v kartoteko bolnikov.
04.5 Interakcije z drugimi zdravili in druge oblike interakcij -
Glede na potencialno občutljivost mieloidnih celic, ki se hitro delijo, na citotoksično kemoterapijo, je treba zdravilo Neulasta dajati vsaj 24 ur po uporabi citotoksične kemoterapije. V kliničnih študijah je bila uporaba zdravila Neulasta 14 dni pred kemoterapijo varna. Uporaba zdravila Neulasta sočasno s katero koli kemoterapijo pri bolnikih ni bila ovrednotena..
Klinične študije niso posebej raziskale možnih interakcij z drugimi hematopoetskimi rastnimi faktorji in citokini.
Možna interakcija z litijem, ki prav tako spodbuja sproščanje nevtrofilcev, ni bila posebej raziskana, ni dokazov, da bi bila ta interakcija lahko škodljiva.
Varnost in učinkovitost zdravila Neulasta nista bili ovrednoteni pri bolnikih, ki so prejemali kemoterapijo, povezano z zapoznelo mielosupresijo, kot so nitro -sečnine.
Specifičnih študij o medsebojnem delovanju ali presnovi niso izvedli; klinične študije pa niso pokazale medsebojnega delovanja zdravila Neulasta z drugimi zdravili.
04.6 Nosečnost in dojenje -
Nosečnost
Podatkov o uporabi pegfilgrastima pri nosečnicah ni ali so omejeni. Študije na živalih so pokazale strupenost za razmnoževanje (glejte poglavje 5.3). Neulasta se ne priporoča med nosečnostjo in pri ženskah v rodni dobi, ki ne uporabljajo kontracepcijskih ukrepov.
Ženske, za katere se ugotovi, da so med zdravljenjem z zdravilom Neulasta noseče, se vabijo, da se vpišejo v program Amgenov nadzor nosečnosti. Kontaktni podatki so navedeni v poglavju 6 Navodila za uporabo.
Čas hranjenja
Podatkov o izločanju zdravila Neulasta / presnovkov v materino mleko ni dovolj. Tveganja za novorojenčke / dojenčke ni mogoče izključiti. Odločiti se je treba o prekinitvi dojenja ali prekinitvi / opustitvi zdravljenja z zdravilom Neulasta ob upoštevanju koristi dojenja. za otroka in koristi zdravljenja za žensko.
Doječe ženske med zdravljenjem z zdravilom Neulasta vabimo, da se vpišejo v Amgenov program spremljanja laktacije. Kontaktni podatki so navedeni v poglavju 6 Navodila za uporabo.
Plodnost
Pegfilgrastim ni vplival na sposobnost razmnoževanja ali plodnost pri samcih ali samicah podgan pri kumulativnem tedenskem odmerku, ki je približno 6 do 9 -krat večji od največjega priporočenega odmerka pri človeku (glede na telesno površino) (glejte poglavje 5.3).
04.7 Vpliv na sposobnost vožnje in upravljanja s stroji -
Neulasta nima vpliva ali ima zanemarljiv vpliv na sposobnost vožnje in upravljanja s stroji.
04.8 Neželeni učinki -
Povzetek varnostnega profila
Najpogosteje poročana neželena učinka sta bili bolečina v kosteh (zelo pogosti [≥ 1/10]) in mišično -skeletni bolečini (pogosti). Bolečine v kosteh so bile na splošno blage do zmerne, prehodne in so bile pri večini bolnikov obvladljive s pogostimi analgetiki.
Pri prvih ali naslednjih odmerkih zdravila Neulasta so poročali o primerih preobčutljivostnih reakcij, vključno s kožnim izpuščajem, urtikarijo, angioedemom, dispnejo, eritemom, zardevanjem in hipotenzijo (občasni [≥ 1 / 1.000, anafilaksa, lahko se pojavijo pri bolnikih, ki prejemajo zdravilo Neulasta (občasno ) (glejte poglavje 4.4).
O sindromu kapilarnega puščanja, ki je lahko smrtno nevaren, če zdravljenje zamuja, so poročali kot o redkih (≥ 1 / 1.000 do
Splenomegalija, običajno asimptomatska, je redka.
Po dajanju pegfilgrastima so poročali o občasnih primerih rupture vranice, vključno s smrtnimi primeri (glejte poglavje 4.4).
Poročali so o občasnih pljučnih neželenih učinkih, vključno z intersticijsko pljučnico, pljučnim edemom, pljučnimi infiltrati in pljučno fibrozo. Občasni primeri so povzročili odpoved dihanja ali sindrom akutne dihalne stiske (Akutni respiratorni distresni sindrom, ARDS), ki so lahko usodni (glejte poglavje 4.4).
Poročali so o posameznih primerih srpastih celičnih kriz (občasni pri takih bolnikih) pri bolnikih s srpasto lastnostjo ali srpastocelično boleznijo (glejte poglavje 4.4).
Tabela neželenih učinkov
Podatki v spodnji tabeli opisujejo neželene učinke, o katerih so poročali v kliničnih študijah in spontanih poročilih. V vsakem frekvenčnem razredu se o neželenih učinkih poroča po padajočem vrstnem redu resnosti.
¹ Glejte poglavje "Opis izbranih neželenih učinkov" spodaj.
² Ta neželeni učinek je bil ugotovljen s postmarketinškim nadzorom, vendar ga v randomiziranih kontroliranih preskušanjih pri odraslih niso opazili. Razred pogostnosti je bil določen s statističnim izračunom na podlagi 1.576 bolnikov, zdravljenih z zdravilom Neulasta v devetih randomiziranih kliničnih preskušanjih.
Opis izbranih neželenih učinkov
Poročali so o občasnih primerih Sweetovega sindroma, čeprav je lahko v nekaterih primerih prispevala osnovna prisotnost hematoloških malignomov.
Pri bolnikih, zdravljenih z zdravilom Neulasta, so poročali o občasnih dogodkih kožnega vaskulitisa. Mehanizem, ki povzroča vaskulitis pri bolnikih, zdravljenih z zdravilom Neulasta, ni znan.
Reakcije na mestu injiciranja, vključno z eritemom na mestu injiciranja (občasni (≥ 1 / 1.000,
Poročali so o pogostih primerih (≥ 1/100, 100 x 109 / l) (glejte poglavje 4.4).
Pri bolnikih, zdravljenih z zdravilom Neulasta po citotoksični kemoterapiji, so reverzibilna, blaga ali zmerna zvišanja, ki jih ne spremljajo klinični simptomi, sečne kisline in alkalne fosfataze občasna; Reverzibilna, blaga ali zmerna zvišanja, ki jih ne spremljajo klinični simptomi, pri laktat dehidrogenazi so redki.
Slabost in glavobol sta bila zelo pogosto opažena pri bolnikih, ki so prejemali kemoterapijo.
Pri bolnikih, ki so po citotoksični kemoterapiji prejemali pegfilgrastim, so opazili občasne primere povišanih testov delovanja jeter (LFT) za ALT (alanin aminotransferazo) ali AST (aspartat aminotransferazo). Ta povečanja so prehodna in reverzibilna.
Poročali so o pogostih primerih trombocitopenije.
V obdobju trženja z uporabo faktorjev, ki stimulirajo kolonije granulocitov, so poročali o primerih sindroma kapilarnega puščanja, ki so se na splošno pojavili pri bolnikih z napredovalo maligno boleznijo, sepso, ki jemljejo več kemoterapevtskih zdravil ali so na aferezi (glejte poglavje 4.4).
Pediatrična populacija
Izkušnje pri otrocih so omejene.Visoke pogostnosti resnih neželenih učinkov so opazili pri otrocih, starih od 0 do 5 let (92%), v primerjavi s starejšimi otroki, starimi od 6 do 11 let, oziroma pri 12 do 21 letih (80%in 67%), in pri odraslih. Najpogostejši prijavljeni neželeni učinek je bila bolečina v kosteh (glejte poglavji 5.1 in 5.2).
Poročanje o domnevnih neželenih učinkih
Poročanje o domnevnih neželenih učinkih, ki se pojavijo po odobritvi zdravila, je pomembno, saj omogoča stalno spremljanje razmerja med koristmi in tveganji zdravila. Zdravstvene delavce prosimo, da o vsakem sumu na neželene učinke poročajo prek nacionalnega sistema za poročanje (Italijanska agencija za zdravila - Spletno mesto: www.agenziafarmaco.gov.it/it/responsabili).
04.9 Preveliko odmerjanje -
Enkraten odmerek 300 mcg / kg je bil subkutano apliciran omejenemu številu zdravih prostovoljcev in pri bolnikih z pljučnim rakom brez mikrocitoma, brez resnih neželenih učinkov. Neželeni učinki so bili podobni tistim pri osebah, ki so prejemale manjše odmerke pegfilgrastima.
05.0 FARMAKOLOŠKE LASTNOSTI -
05.1 "Farmakodinamične lastnosti -
Farmakoterapevtska skupina: imunostimulansi, faktor, ki stimulira kolonije.
Oznaka ATC: L03AA13.
Faktor stimulacije kolonije človeških granulocitov (G-CSF) je glikoprotein, ki uravnava proizvodnjo in sproščanje nevtrofilcev iz kostnega mozga. Pegfilgrastim je sestavljen iz rekombinantne humane molekule G-CSF (r-metHuG-CSF), kovalentno povezane z eno samo 20 kd molekulo polietilen glikola (PEG). Pegfilgrastim je dolgotrajna oblika filgrastima zaradi zmanjšanega ledvičnega očistka. Pegfilgrastim in filgrastim imata enak mehanizem delovanja, kar povzroči znatno povečanje števila perifernih nevtrofilcev v 24 urah z zanemarljivim povečanjem monocitov in / ali limfocitov. Podobno kot filgrastim imajo nevtrofilci, ki nastanejo kot odziv na pegfilgrastim, normalno ali povečano delovanje, kar dokazujejo ocene kemotaktične in fagocitne aktivnosti. Tako kot drugi hematopoetski rastni faktorji je G-CSF pokazal in vitro stimulativne lastnosti na človeških endotelijskih celicah. G-CSF lahko spodbuja rast in vitro mieloidnih celic je mogoče odkriti celo maligne in podobne učinke in vitro na nekaterih ne-mieloidnih celicah.
V dveh randomiziranih, dvojno slepih, ključnih študijah pri bolnikih z visokorizičnim rakom dojke II-IV stopnje na mielosupresivni kemoterapiji, vključno z doksorubicinom in docetakselom, je uporaba pegfilgrastima kot enkratnega odmerka enkrat na cikel skrajšala trajanje nevtropenije in incidenco febrilne nevtropenije, podobne tisti, ki jo opazimo pri dnevnem odmerjanju filgrastima (mediana 11 odmernih dni). Ker ni bilo podpore rastnega faktorja, so poročali, da je ta vzorec povzročil povprečno trajanje nevtropenije 4. stopnje 5-7 dni z incidenco febrilne nevtropenije 30-40%. V eni študiji (n = 157) z uporabo 6 mg fiksnega odmerka pegfilgrastima, je bilo povprečno trajanje nevtropenije 4. stopnje v skupini s pegfilgrastimom 1,8 dni v primerjavi s 1,6 dni v skupini s filgrastimom (razlika 0,23 dni, 95% IZ: -0,15, 0,63). je bila stopnja vročinske nevtropenije 13% bolnikov, zdravljenih s pegfilgrastimom, v primerjavi z 20% bolnikov, zdravljenih s filgrastimom (razlika 7%, 95% IZ: - 19%, 5%). V drugi študiji (n = 310) je bilo z uporabo odmerka, prilagojenega telesni masi (100 mcg / kg), povprečno trajanje nevtropenije 4. stopnje v skupini s pegfilgrastimom 1,7 dni v primerjavi z 1,8 dni v skupini s filgrastimom (razlika 0,03 dni, 95% IZ: -0,36, 0,30). Skupna stopnja febrilne nevtropenije je bila 9% bolnikov, zdravljenih s pegfilgrastimom, in 18% bolnikov, zdravljenih s filgrastimom (razlika 9%, 95% IZ: -16,8%, -1,1%).
V dvojno slepi, s placebom kontrolirani študiji pri bolnikih z rakom dojke je bil učinek pegfilgrastima na pojavnost vročinske nevtropenije ocenjen po dajanju režima kemoterapije, povezanega z 10–20-odstotno pojavnostjo febrilne nevtropenije (docetaksel 100 mg / m² vsake 3 tedne po 4 cikle) .Dvajset osemindvajset bolnikov je bilo naključno izbranih za prejemanje enkratnega odmerka pegfilgrastima ali placeba približno 24 ur po kemoterapiji v vsakem ciklu (2. dan). Incidenca febrilne nevtropenije je bila pri randomiziranih bolnikih nižja. prejemali pegfilgrastim v primerjavi s placebom (1% proti 17%, str
Omejen vzorec (n = 83), randomizirana, dvojno slepa študija druge faze, izvedena pri bolnikih na kemoterapiji zaradi akutne mieloične levkemije de novo primerjali pegfilgrastim (enkratni odmerek 6 mg) s filgrastimom med indukcijsko kemoterapijo. Mediana časa do remisije hude nevtropenije je bila v obeh zdravljenih skupinah 22 dni. Dolgoročnega izida niso preučevali (glejte poglavje 4.4).
V multicentrični, randomizirani, odprti študiji faze II (n = 37) pri pediatričnih bolnikih s sarkomom, ki so po prvem tečaju kemoterapije z vinkristinom, doksorubicinom in ciklofosfamidom (VAdriaC / IE) prejeli 100 mcg / kg pegfilgrastima daljše trajanje hude nevtropenije (nevtrofilci
05.2 "Farmakokinetične lastnosti -
Največja serumska koncentracija pegfilgrastima je opažena 16 do 120 ur po dajanju enkratnega podkožnega odmerka; serumske koncentracije v obdobju nevtropenije po mielosupresivni kemoterapiji ostanejo stabilne. Izločanje pegfilgrastima je nelinearno glede na odmerek; serumski očistek pegfilgrastima se zmanjšuje s povečanjem odmerka. Zdi se, da se pegfilgrastim izloča predvsem z očistkom, ki ga posredujejo nevtrofilci, ki je pri večjih odmerkih nasičen. V skladu s samoreguliranim mehanizmom očistka se serumska koncentracija pegfilgrastima hitro znižuje skupaj z naraščanjem nevtrofilcev.
Zaradi mehanizma očistka, ki ga posredujejo nevtrofili, ni pričakovati, da bi okvara jeter ali ledvic vplivala na farmakokinetiko pegfilgrastima. V odprti študiji z enim odmerkom (n = 31) različne stopnje okvare ledvic, vključno s končno stopnjo ledvične bolezni, niso vplivale na farmakokinetiko pegfilgrastima.
Starejša populacija
Omejeni razpoložljivi podatki kažejo, da je farmakokinetika pegfilgrastima pri starejših osebah (> 65 let) podobna kot pri odraslih.
Pediatrična populacija
Farmakokinetiko pegfilgrastima so proučevali pri 37 pediatričnih bolnikih s sarkomom, ki so po zaključku kemoterapije VAdriaC / IE prejemali 100 μg / kg pegfilgrastima. Mlajša starostna skupina (0-5 let) je imela višjo "povprečno izpostavljenost pegfilgrastimu (AUC) (± standardni odklon) (47,9 ± 22,5 mcg • h / ml) kot otroci, starejši od 6-11 let in 12-21 let (22,0 ± 13,1 mcg • hr / ml oziroma 29,3 ± 23,2 mcg • hr / ml) (glejte poglavje 5.1).
Z izjemo mlajše starostne skupine (0-5 let) je bila povprečna AUC pri pediatričnih bolnikih podobna kot pri odraslih bolnikih z visokorizičnim rakom dojke II-IV stopnje, ki so po koncu doksorubicina prejele 100 mcg / kg pegfilgrastima / docetaksel (glejte poglavji 4.8 in 5.1).
05.3 Predklinični podatki o varnosti -
Predklinični podatki iz tradicionalnih študij toksičnosti pri ponavljajočih se odmerkih so pokazali pričakovane farmakološke učinke, vključno s povečanjem števila belih krvnih celic, mieloidno hiperplazijo kostnega mozga, ekstramedularno hematopoezo in splenomegalijo.
Pri podganah, ki so se rodile pri brejih samicah, katerim so subkutano dajali pegfilgrastim, niso opazili škodljivih učinkov, pri kuncih pa je subkutano dajanje pegfilgrastima povzročilo toksičnost zarodka in ploda (izguba zarodka) v kumulativnih odmerkih 4-kratnega priporočenega odmerka za ljudi. Študije na podganah so pokazale, da je možen transplacentarni prehod pegfilgrastima. Študije na podganah so pokazale, da subkutana uporaba pegfilgrastima ni vplivala na sposobnost razmnoževanja, plodnost, cikel estrusa, dneve med parjenjem in spolnim odnosom ter intrauterino preživetje. Pomen teh podatkov za ljudi ni znan.
06.0 FARMACEVTSKE INFORMACIJE -
06.1 Pomožne snovi -
Natrijev acetat *
Sorbitol (E420)
Polisorbat 20
Voda za injekcije
* Natrijev acetat dobimo s titracijo ledene ocetne kisline z natrijevim hidroksidom.
06.2 Nezdružljivost "-
Tega zdravila ne smete mešati z drugimi zdravili, zlasti z raztopinami natrijevega klorida.
06.3 Obdobje veljavnosti "-
3 leta.
06.4 Posebna navodila za shranjevanje -
Shranjujte v hladilniku (2 ° C - 8 ° C).
Zdravilo Neulasta lahko hranite pri sobni temperaturi (ne nad 30 ° C) enkrat in največ 72 ur. Zdravilo Neulasta, ki ga pustite pri sobni temperaturi več kot 72 ur, je treba zavreči.
Ne zamrzujte. Nenamerna izpostavljenost nizkim temperaturam, enkrat manj kot 24 ur, ne vpliva na stabilnost zdravila Neulasta.
Vsebnik shranjujte v zunanji ovojnini, da zdravilo zaščitite pred svetlobo.
06.5 Vrsta neposredne embalaže in vsebina pakiranja -
Napolnjena injekcijska brizga (steklo tipa I) z gumijastim batom in iglo iz nerjavečega jekla z ali brez avtomatskega ščitnika za iglo.
Pokrovček igle napolnjene injekcijske brizge vsebuje suh naravni kavčuk (derivat lateksa) (glejte poglavje 4.4).
Vsaka napolnjena injekcijska brizga vsebuje 0,6 ml raztopine za injiciranje. Velikost pakiranja ene napolnjene injekcijske brizge, pakirana z pretisnim omotom ali brez pretisnega omota.
Na trgu ni vseh navedenih pakiranj.
06.6 Navodila za uporabo in ravnanje -
Pred dajanjem raztopine zdravila Neulasta je treba preveriti odsotnost vidnih delcev, injicirati je treba le bistro in brezbarvno raztopino.
Če se pegfilgrastim prekomerno vznemirja, lahko tvori agregate in postane biološko neaktiven.
Pred injiciranjem raztopine pustite, da napolnjena injekcijska brizga doseže sobno temperaturo.
Neuporabljeno zdravilo in odpadke, pridobljene iz tega zdravila, je treba odstraniti v skladu z lokalnimi predpisi.
07.0 IMETNIK "DOVOLJENJA ZA PROMET" -
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
Nizozemska
08.0 ŠTEVILKA DOVOLJENJA ZA PROMET -
EU/1/02/227/001 Injekcijska brizga z 1 pakiranjem z pretisnim omotom
035716012
EU/1/02/227/002 1 pakiranje brizge brez pretisnega omota
EU/1/02/227/004 Injekcijska brizga z 1 pakiranjem z pretisnim omotom z ščitnikom za iglo
035716036
09.0 DATUM PRVEGA DOVOLJENJA ALI PODALJŠANJA DOVOLJENJA -
Datum prve odobritve: 22. avgust 2002
Datum zadnje obnove: 16. julij 2007
10.0 DATUM REVIZIJE BESEDILA -
Maja 2015